| Identification | Back Directory | [Name]
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[8.7.6]tricos-11-ene-2,5,8,19,22-pentone | [CAS]
128517-07-7 | [Synonyms]
FK228
CS-972
istodax
Chromadax
Romidisin
FR 901228
Romidepsin
NSC 630176
depsipeptide
5)-disulfide
RoMidepsin-d8
RoMidepsin(FK228)
Romidepsin, >=98%
FK228 RoMidepsin
Antibiotic FR 901228
RoMidepsin (FK228 ,Depsipeptide)
Romidepsin
Antibiotic FR 901228
Romidepsin (FK228, Depsipeptide, FR1228)
RoMidepsin/Depsipeptide(FK-228, NSC-63017)
Romidepsin, 98%, a potent HDAC1 and HDAC2 inhibitor
FK 228;? FR 901228;? NSC 630176;? DEPSIPEPTIDE;FK228
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[
[S-(E)]-N-(3-Hydroxy-7-Mercapto-1-oxo-4-
heptenyl)-D-valyl-D-cysteinyl-(Z)-2,3-didehydro-2-aMinobutanoyl-
Cyclo[(2Z)-2-aMino-2-butenoyl-L-valyl-(3S,4E)-3-hydroxy-7-Mercapto-4-heptenoyl-D-valyl-D-cysteinyl],cyclic (3®
Cyclo[(2Z)-2-aMino-2-butenoyl-L-valyl-(3S,4E)-3-hydroxy-7-Mercapto-4-heptenoyl-D-valyl-D-cysteinyl] Cyclic (35)-Disulfide
Cyclo[(2Z)-2-aMino-2-butenoyl-L-valyl-(3S,4E)-3-hydroxy-7-Mercapto-4-heptenoyl-D-valyl-D-cysteinyl]-d8 Cyclic (35)-Disulfide
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[8.7.6]tricos-11-ene-2,5,8,19,22-pentone
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[8.7.6]tricos-11-ene-2,5,8,19,23-pentone
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[8.7.6]tricos-11-ene-2,5,8,19,22-pentone USP/EP/BP
(1S,4Z,7S,10S,11E,20R)-4-ethylidene-7,20-dipropan-2-yl-9-oxa-15,16-dit hia-3,6,18,21-tetrazabicyclo[8.7.6]tricos-11-ene-2,5,8,19,22-pentone RoMidepsin (FK228, Depsipeptide) | [Molecular Formula]
C24H36N4O6S2 | [MDL Number]
MFCD06199142 | [MOL File]
128517-07-7.mol | [Molecular Weight]
540.7 |
| Chemical Properties | Back Directory | [Melting point ]
219-224°C | [Boiling point ]
942.8±65.0 °C(Predicted) | [density ]
1.174 | [storage temp. ]
-20°C | [solubility ]
Soluble in DMSO (up to 10 mg/ml). | [form ]
powder | [pka]
11.33±0.60(Predicted) | [color ]
white to beige | [Stability:]
Stable for 2 years from date of purchase as supplied. Solutions in DMSO may be stored at -20° for up to 1 month. |
| Safety Data | Back Directory | [RTECS ]
YV9399000 | [HS Code ]
29349990 | [Toxicity]
mouse,LD50,intraperitoneal,6400ug/kg (6.4mg/kg),Journal of Antibiotics. Vol. 47, Pg. 301, 1994. |
| Hazard Information | Back Directory | [Description]
The U.S. FDA approved romidepsin (also referredto as FK228) in 2009 for
the treatment of cutaneous T-cell lymphoma (CTCL) for patients who
received at least one systemic therapy.
Romidepsin is a natural product that was
first isolated from the fermentation broth of C. violaceum. Romidepsin is the
second histone deacetylase (HDAC) inhibitor approved for CTCL, the other
being vorinostat,whichwas approved by the FDA in 2006. Unlike vorinostat
which is a pan-HDAC inhibitor, romidepsin shows modest selectivity for
class I HDACs in in vitro assays. It has been shown that after romidepsin
enters the cytoplasm, the disulfide bond is cleaved by glutathione to release
the sulfhydryl groupwhich chelateswith the activesite zinc of class IHDACs
and inhibits the enzymatic activity at nanomolar concentrations.
Although romidepsin inhibits class I HDACs, it is 17–23 times less potent as
the parent than the corresponding reduced form at each isozyme. For example,
the IC50 of romidepsin at HDAC1 is 36±16nM while that of the reduced form is IC50=1.6± 0.9nM.Romidepsinhasalsobeenshownto induce cell cycle arrest, differentiation, and apoptosis in tumor cells by mechanisms
that cannot be completely explained by HDAC inhibition alone. | [Chemical Properties]
Pale Yellow Solid | [Originator]
Fujisawa (Astellas Pharma) (Japan) | [Uses]
Labelled Romidepsin (R425060). Romidepsin is a histone deacetylase inhibitor that can alter chromatin structure and gene transcription leading to multiple changes in cellular protein production. This
may result in cell cycle arrest and tumor growth inhibition. Romidepsin has shown anti-proliferative activity in vitro against multiple mouse and human tumor cell lines and in vivo in human tumor xeno
graft models. Romidepsin can be administered with a second agent, such as a cytotoxic agent, a steroidal agent, a proteasome inhibitor, or a kinase inhibitor. | [Definition]
ChEBI: A cyclodepsipeptide consisting of the cyclic disulfide of (2Z)-2-aminobut-2-enoyl, L-valyl, (3S,4E)-3-hydroxy-7-sulfanylhept-4-enoyl, D-valyl and D-cystei
yl residues coupled in sequence and cyclised head-to tail. | [Brand name]
Chromadax (Gloucester);Istodax. | [Biochem/physiol Actions]
Romidepsin is a very potent natural prodrug inhibitor of HDAC1 and HDAC2 that is converted to active form by glutathione. Romidepsin has IC50 values of 36 nM and 47 nM for HDAC1 and HDAC2, respectively. Romidepsin kills lymphoma cell lines overexpressing Bcl-2 and Bcl-XL, and has been approved for the treatment for cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma, and a variety of other cancers. | [Clinical Use]
Romidepsin, a histone deacetylase inhibitor, originally developed by Fujisawa (now Astellas
Pharma), causes cell cycle arrest, differentiation, and apoptosis in various cancer cells. In 2004,
the FDA granted fast-track designation for romidepsin as monotherapy for the treatment of cutaneous Tcell
lymphoma (CTCL) in patients who have relapsed following, or become refractory to, other systemic
therapies. The FDA designated romidepsin as an orphan drug and it was approved in 2009 for this
indication and it was commercialized in 2010. In 2007, another fast-track designation was granted for
the product as monotherapy of previously treated peripheral T-cell lymphoma. Romidepsin (FR901228)
was originally discovered and isolated from the fermentation broth of Chromobacterium violaceum No.
968. It was identified through efforts in the search for novel agents which selectively reverse the
morphological phenotype of Ras oncogene-transformed cells since the Ras signaling pathway plays a
critical role in cancer development. Therefore, the drug could also have multiple molecular targets for
its anticancer activity besides HDAC. FR901228 is a bicyclic depsipeptide which is structurally
unrelated to any known class of cyclic peptides with an unusual disulfide bond connecting a thiol and Dcysteine. | [Synthesis]
Romidepsin is commercially produced by fermentation; however its interesting and novel
structure warrants examination of its synthesis within the context of this review. The
synthesis of romidepsin described is based on the total synthesis reported by the Williams and
Simon groups. L-Valine methyl ester (134) was coupled to N-Fmoc-L-threonine in
the presence of the BOP reagent in 95% yield. The N-Fmoc protecting group was removed with Et2NH
and the corresponding free amine was coupled to N-alloc-(S-triphenylmethyl)-D-cysteine with 1-ethyl-3-
(3-dimethylaminopropyl)carbodiimide (EDCI) and HOBT in DMF and CH2Cl2 to yield the tripeptide
135 in good yield. The threonine residue of tripeptide 135 was then subjected to dehydrating conditions
to give alkene 136 in 95% yield. The N-alloc protecting group of the dehydrated tripeptide 136 was
removed with palladium and tin reagents and the corresponding free amine was subsequently coupled with N-Fmoc-D-valine to give tetrapeptide 137 in 83% yield. After removal of the N-Fmoc protecting
group of compound 137 with Et2NH amine 138 was obtained in quantitative yield. The acid coupling
partner 145 for amine 138 was prepared as follows: methyl 3,3-dimethoxypropionate (139) was
converted to its corresponding Weinreb amide by standard conditions and reacted with lithium acetylide
140 to give propargylic ketone 141 in 75% yield. Noyori?ˉs asymmetric reduction of ketone 141 using
ruthenium catalyst 142 gave the (R)-propargylic alcohol in 98% ee. This was followed by Red-Al
reduction of the alkyne to selectively yield (E)-alkene 143 in 58% yield for the two steps. Liberation of
the primary alcohol with tetrabutylammonium fluoride (TBAF) followed by selective tosylation gave
144 in 70% yield in two steps. Hydrolysis of the dimethyl acetal of 144 with LiBF4 was followed by a
Pinnick oxidation to give the corresponding carboxylic acid. The tosylate was displaced with trityl
mercaptan in the presence of tert-butyl alcohol to give allylic alcohol 145 in 65% yield for the three
steps. Aminoamide 138 was then coupled to acid 145 using BOP to give peptide 146 in quantitative
yield. The methyl ester of compound 146 was hydrolyzed with lithium hydroxide to provide the free
carboxylic acid which underwent macrolactonization under Mitsunobu conditions in the presence of
diisopropyl azodicarboxylate (DIAD) and triphenylphosine to give macrocycle 147 in 24% yield.
Finally, the disulfide linkage was formed by treating bis-tritylsulfane 147 with iodine in methanol at
room temperature to give romidepsin (XIII) in 81% yield.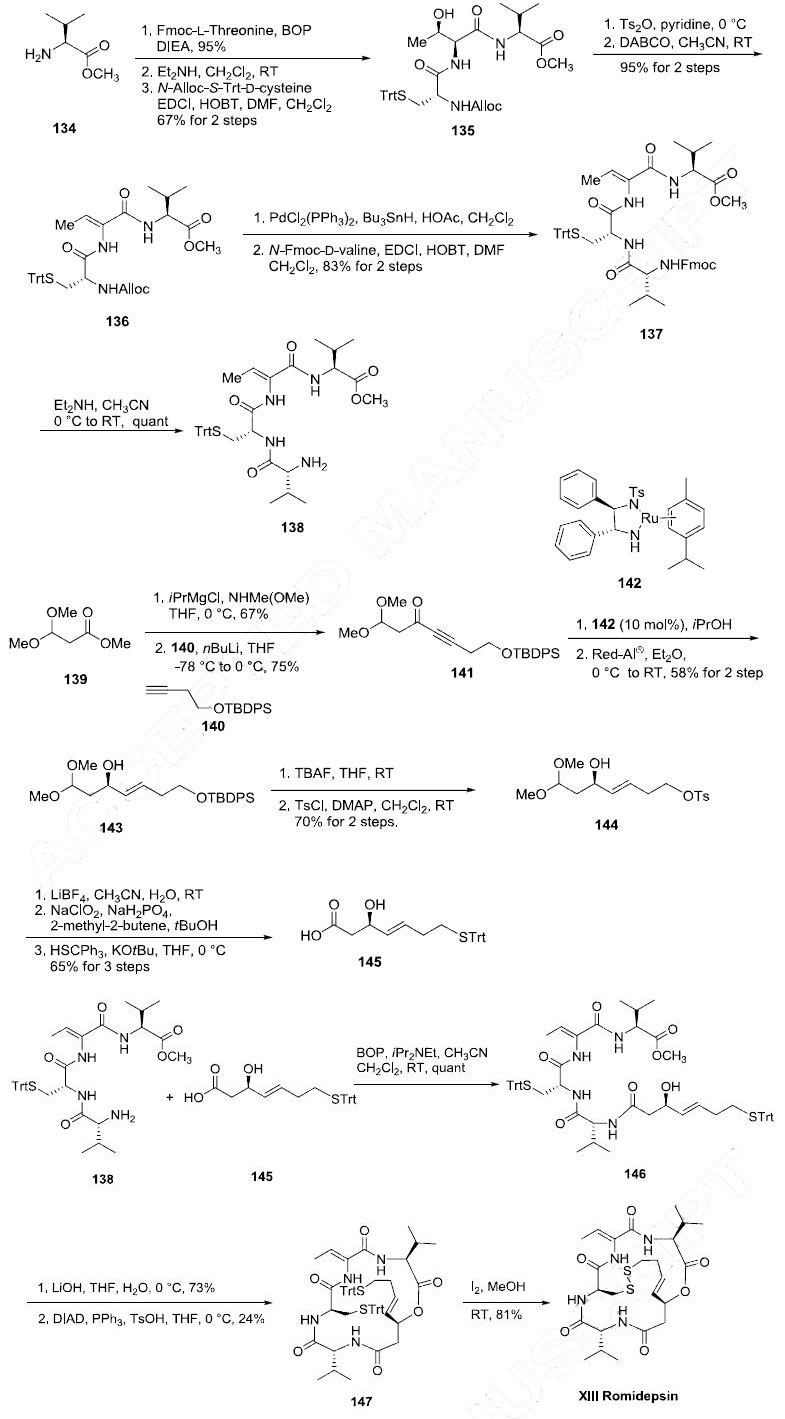
| [storage]
Store at -20°C,unstable in solution, ready to use. | [References]
1) Furumai et al. (2002), FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases; Cancer Res., 62 4916
2) Panicker et al. (2010), Romidepsin (FK228/depsipeptide) controls growth and induces apoptosis in neuroblastoma tumor cells; Cell Cycle, 9 1830
3) Ueda et al. (1994), FR901228, a novel anti-tumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice; J. Antibiot. (Tokyo), 47 315
4) VanderMolin et al. (2011), Romodepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma; J. Antibiotic. (Tokyo), 64 525 |
|
|