1352792-74-5
 1352792-74-5 结构式
1352792-74-5 结构式
基本信息
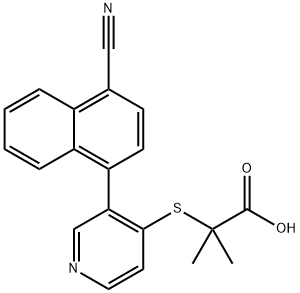
2-((3-(4-氰基萘-1-基)吡啶-4-基)硫基)-2-甲基丙酸
HY-16733
RDEA3170
Verinurad
Verinurad (RDEA3170)
2-((3-(4-cyanonaphthalen-1-yl)pyridin-4-yl)thio)-2-methylpropanoic acid
2-[[3-(4-Cyano-1-naphthalenyl)-4-pyridinyl]thio]-2-methylpropanoic acid
Propanoic acid, 2-[[3-(4-cyano-1-naphthalenyl)-4-pyridinyl]thio]-2-methyl-
物理化学性质
常见问题列表
维立诺雷属于小分子化合物,是有机阴离子转运蛋白URAT1(SLC22A12)抑制剂,可用于治疗痛风和高尿酸血症。
| Target | Value |
|
URAT1 transporter
(Cell-free assay) | 25 nM |
Verinurad以浓度依赖性方式抑制人源URAT1的转运活性,IC50为25 nM。Verinurad还抑制URAT1相关同系物OAT4和OAT1,IC50分别为5.9 μM和4.6 μM,亲和力较之与URAT低200倍。Verinurad对人源URAT1比对大鼠URAT1的亲和力高1640倍(human URAT1 IC50=0.025 μM,rat URAT IC50=41 μM)。Verinurad对人源URAT1具有高效力,URAT1的第35、365和481位残基有助于verinurad结合。在人源URAT1在365位引入一个嵌合点突变(人源365位苯丙氨酸替换成大鼠365位的酪氨酸;h-F365Y),verinurad对其IC50为4 μM,比对野生型人源URAT1D低160倍。同时,如果在输入URAT1引入类似的点突变(即将大鼠365位酪氨酸突变为人源365位的苯丙氨酸;r-Y365F),verinurad对其IC50为2.9 μM,比对野生型大鼠URAT1高14倍。
在人体中,Verinurad单药给药吸收很快,其暴露量(Cmax和AUC)随剂量的升高而升高。在饥饿状态下,给药0.5-0.75小时后达到Cmax;在进食状态下,给药后1.25小时达到Cmax。在不同剂量下,t 1/2 约为10-15小时。Verinurad在体内耐受性良好。在体循环中,verinurad具有高清除率(CL/F ranged ~30-50 L/h),广泛分布在血管外。其肾排泄率低,在尿液中仅2%-3%。大部分verinurad可能在肝脏中通过生物转化形成代谢物。