国家食品药品监督管理总局药品审评中心于2016年4月20日公布了双鹭药业的Lenalidomide(来那度胺)为“临床急需或与我国现有治疗药品比较具有明显的临床优势,拟纳入优先审评程序”。这一消息在业界引发热议,本期编者将通过双鹭药业与Clegene公司所申请的专利,对比分析这两家公司的合成工艺路线。
背景介绍:
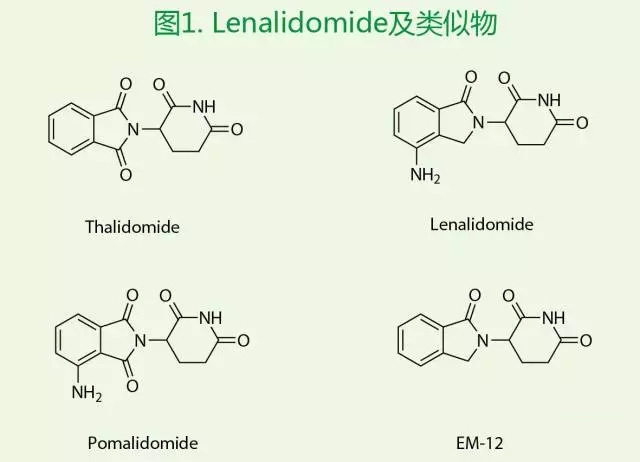
Lenalidomide(CDC-501和CC-5013),美国Clegene公司研发的三大明星药物之一(Thalidomide,Lenalidomide和Pomalidomide,图1),属于TNF-α抑制剂,具有免疫调节,抗血管生成和抗肿瘤特性。首先于2005年12月27日经FDA批准在美国上市,之后分别在欧洲(2007年6月14日)、日本(2010年6月25日)和中国(2013年1月23日)上市,用于治疗骨髓增生/发育异常综合征,套细胞淋巴瘤和多发性骨髓瘤。美国FDA和欧盟EMA又分别在2015年2月17日和2月20日相继扩展了Lenalidomide的治疗适应症——和地塞米松联合使用作为一线用药治疗多发性骨髓瘤。根据Celgene公司年报显示,Lenalidomide 2014年全球销售额达到49.8亿美元,2015年销售总额58亿美元,增长率为16.5%,其全球年销售额预计5年内将再翻一番,至2020年销售总额或高达100亿美元。
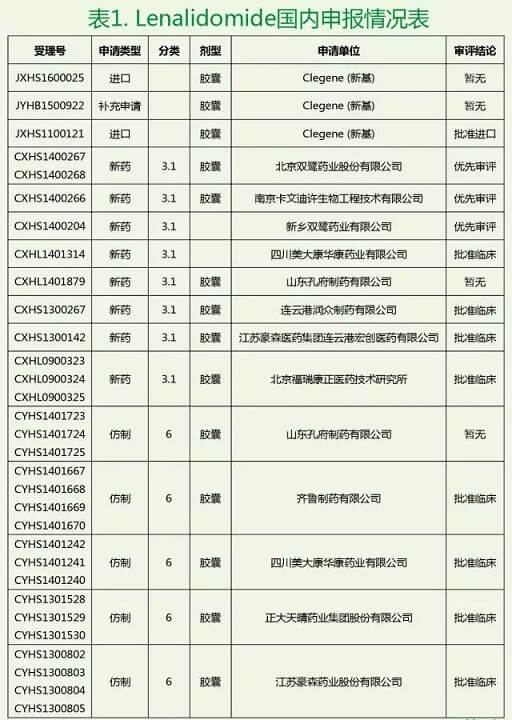
双鹭药业的Lenalidomide制剂已经完成了验证性临床研究,于2014年11月18日进入CDE进行审评,是国内家申请生产的。2016年4月20日又获得优先审评权,将很快在国内上市。目前,国内除美国Celgene公司的进口产品在销售及双鹭药业获得优先审评外,还有其他制药企业申请报批如表1所示。
药物发现:
Lenalidomide是Thalidomide(沙利度胺)类似物,其药物发现需要从Thalidomide说起。Thalidomide是一个消旋体,“反应停”事件发现(S)-Thalidomide是引起致畸性的元凶,但是研究表明(R)-Thalidomide在体内很容易发生消旋,对于孕妇服用带来的致畸性不可避免,1961年11月被强行撤市。但是,科学家并未全盘否定这个药物,而是进行深入研究,特别是在免疫、抗炎、抗血管生成及一些疑难杂症上的临床治疗研究。1964年, Thalidomide被发现能够治疗结节性红斑(ENL);1991年,其抗炎机制被证实,Thalidomide能够治疗ENL主要原因是选择性抑制人单核巨噬细胞受到免疫刺激所释放的肿瘤坏死因子 α (TNF-α )。1998年7月美国生物制药企业Clegene获得FDA批准将Thalidomide用于治疗ENL的功用在美国重新上市。
基于Thalidomide的临床活性研究,化学结构和生理效果,Clegene也在研究开发新颖的类似物或者衍生物,期望去除无法避免的胚胎致畸性。于是对Thalidomide进行了结构改造,合成了氨基邻苯二甲酰基类似物并进行了活性测试。与此同时还测试了异吲哚酮类似物EM-12,发现该化合物具有相同的TNF-α的抑制活性。在后续开发中,考虑到异吲哚酮结构的稳定性要优于邻苯二甲酰胺环,并有可能提高生物利用度,所以基于EM-12的结构优化,发现了对兔子胚胎没有致畸性的Lenalidomide。
合成路线:
鉴于Clegene公司没有申请化合物专利,通过对已公开专利的分析,双鹭药业子公司南京卡文迪许生物工程技术有限公司从设计新颖路线和发明新的晶型出发,发明了一条全新路线进行Lenalidomide的合成并申请专利。除获得国内专利授权外,双鹭药业的Lenalidomide已在美国、欧洲、韩国、日本、澳大利亚、印度等国家申请了专利,部分专利已在美国、欧洲、韩国、日本、澳大利亚获得专利授权。下面编者重点比较一下两家的合成路线。
Lenalidomide的分子结构共包含两个部分,分别是异吲哚酮和2,6-哌啶二酮结构。而专利报道合成路线的区别是异吲哚酮环和哌啶环的关环步骤。
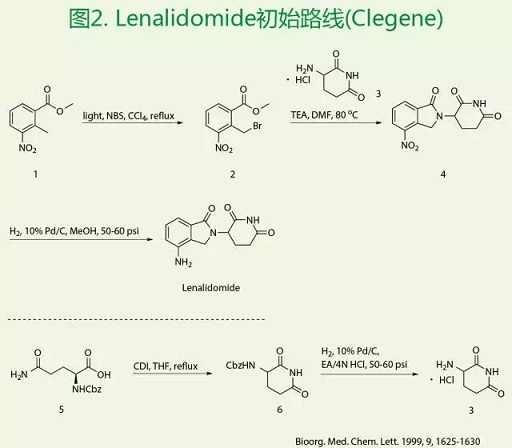
首先是Clegene的初始路线,如图2所示。该路线的设计是先构建哌啶环,再与2-溴甲基-3-硝基苯甲酸甲酯2反应构建哌啶环得到化合物4,然后催化氢化得到Lenalidomide。其中,反应的关键中间体2的合成是以2-甲基-3-硝基苯甲酸甲酯1为原料,CCl4为溶剂,紫外光照条件催化溴化得到。另外一个关键中间体α-氨基戊二酰亚胺盐酸盐3的合成是以N-苄氧羰基-L-谷氨酰胺为起始原料,CDI和THF条件下关环,再催化氢化得到。该方法的缺点:(1)合成化合物2,紫外光照射下催化反应回流反应时间长收率低且工业化困难,对生产人员工作中对催化光源汞灯产生的紫外光的防护比较困难。此外,CCl4毒性大,对臭氧层破坏作用严重,现为联合国环境总署禁止使用的化学品,工业生产时处理困难,对环境极为不利。(2)合成关键中间体3,CDI和THF回流反应时间长,高温回流使得产物纯度偏低,分离纯化困难;催化氢化反应的加压使工业化操作时的危险程度大大增加。
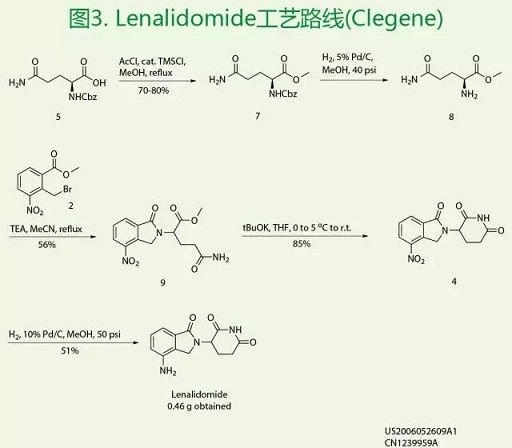
然后是Lenalidomide的工艺路线,如图3所示。2-溴甲基-3-硝基苯甲酸甲酯2与谷胺酰胺甲酯7在碱性条件关环得到关键中间体异吲哚酮8,随后经过两种策略不同顺序关环构建哌啶环合成Lenalidomide。该专利保护合成路线的优点是该路线以天然氨基酸为起始原料,价格低廉。缺点是(1)对反应产物的分离提纯至少二次或者二次以上用到柱层析,工业化操作复杂困难,不利于工业化放大生产。(2)在合成中,两次用到加压催化氢化,导致工业化操作时的危险程度大大增加。(3)整条合成路线的总收率不足20%。
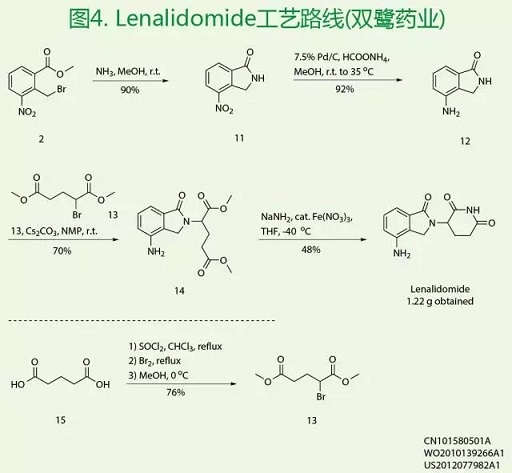
如图4所示,双鹭药业设计一条不同于原研公司的新路线,一定程度上克服了现有技术上存在的不足。首先是以化合物2为起始原料构建异吲哚酮12,再与α-溴代戊二酸二甲酯13发生亲核取代得到化合物14,最后在氨基钠条件下环合构建哌啶环得到Lenalidomide。
该工艺路线与原研公司路线相比,有以下几个优点:(1)合成路线简短,方法简便;且所需的关键底物的制备方法简单,价格低廉,适于工业化生产的要求。(2)反应条件较为温和,无需苛刻的长时间回流反应;且各步所用溶剂易于环保处理,对环境友好。(3)反应所得的产物纯度高,分离提纯简单,无需柱层析等复杂的分离提纯条件。(4)各步反应收率较高,总收率为28%。但是,缺点是路线中涉及不易制备和储存的氨基钠为反应试剂,且起始原料也需要前期合成,也将在一定程度上制约该路线大量制备的有效性。
小结:
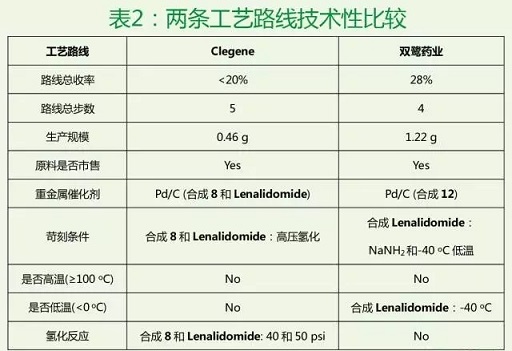
通过两条工艺路线的技术性比较(表2),我们很容易发现这两条路线的优缺点。比如,双鹭药业的工艺路线虽然避免了加压催化氢化,但是在最后合成Lenalidomide这一步采用自制氨基钠及不易控制的低温条件,也存在着一定的不足。著名药物化学家Jie Jack Li有本书叫《The Art of Drug Synthesis》,阐明路线设计和反应条件选取是一门艺术。如何更好地设计一条合成路线,是我们一直探索和追求的。