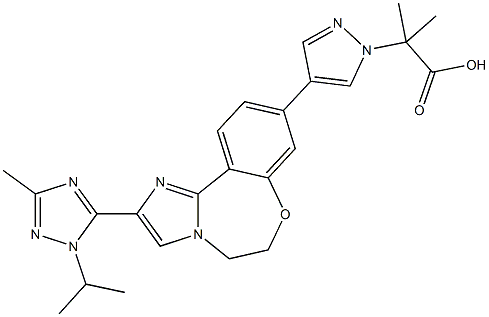
1H-Pyrazole-1-acetic acid, 4-[5,6-dihydro-2-[3-Methyl-1-(1-Methylethyl)-1H-1,2,4-triazol-5-yl]iMidazo[1,2-d][1,4]benzoxazepin-9-yl]-α,α-diMethyl- synthesis
- Product Name:1H-Pyrazole-1-acetic acid, 4-[5,6-dihydro-2-[3-Methyl-1-(1-Methylethyl)-1H-1,2,4-triazol-5-yl]iMidazo[1,2-d][1,4]benzoxazepin-9-yl]-α,α-diMethyl-
- CAS Number:1282513-03-4
- Molecular formula:C24H27N7O3
- Molecular Weight:461.52
![1H-Pyrazole-1-acetic acid, 4-[5,6-dihydro-2-[3-Methyl-1-(1-Methylethyl)-1H-1,2,4-triazol-5-yl]iMidazo[1,2-d][1,4]benzoxazepin-9-yl]-α,α-diMethyl-, ethyl ester](/CAS/GIF/1282514-64-0.gif)
1282514-64-0
12 suppliers
inquiry
1282513-03-4
16 suppliers
inquiry
Yield:1282513-03-4 95%
Reaction Conditions:
Stage #1: ethyl 2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]-oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanoatewith water;lithium hydroxide at 75; for 4.5 h;
Stage #2: with hydrogenchloride in water at 15; pH=1;
Steps:
17 Example 17 2-(4-(2-( 1 -isopropyl-3-methyl- 1 H- 1 ,2,4-triazol-5-yl)-5 ,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoic acid II
The ester saponification reaction was initiated with the addition of 3.5 M aqueous LiOH (0.74 kg in 5.0 L, 17.64 mol, 5 equiv) to the reaction mixture from Example 16 and allowed to warm to 75 °C. The mixture was sampled every 30 min (IPC method: XTerra MS Boronic) and the saponification was complete after 4.5 h (with less than 0.3% 23 remaining). The reaction mixture was concentrated via distillation to approximately half volume (starting vol = 37 L; final vol = 19 L) to remove EtOH and THF, resulting in tan-brown slurry. Water (5 L, 5 vol) was charged to the mixture and then distilled (starting vol = 25 L; final vol = 21 L). The temperature was set at 60 °C (jacket control) and then charged with isopropyl acetate, IP Ac (4 L, 4 vol). The biphasic mixture was stirred a minimum of 5 min and then the layers allowed to separate for a minimum of 5 min. The bottom aqueous layer was removed into a clean carboy and the organics were collected into a second carboy. The extraction process was repeated a total of four times, until the organic layer was visibly clear. The aqueous mixture was transferred back to the reactor and then cooled to 15 °C. A 6 M solution of HC1 (6.4 L, 38.40 mol, 10 equiv) was charged slowly until a final pH = 1 was obtained. The heterogeneous mixture was then filtered. The resulting solids were washed twice with 5 L (2 x 5 vol) of water. The filter was then heated to 80 °C and the vacuum set to -10 Psi (with nitrogen bleed) and the solids were dried for 24 h (KF = 2.0 % H20) to give 1.54 kg (95% corrected yield) of II as a white solid; 98% wt, 97.3 % pure.
References:
WO2014/140073,2014,A1 Location in patent:Page/Page column 14; 29
![IMidazo[1,2-d][1,4]benzoxazepine, 9-broMo-5,6-dihydro-2-[3-Methyl-1-(1-Methylethyl)-1H-1,2,4-triazol-5-yl]-](/CAS/20150408/GIF/1282514-63-9.gif)
1282514-63-9
24 suppliers
inquiry
1282513-03-4
16 suppliers
inquiry
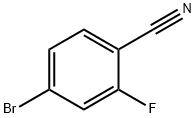
105942-08-3
368 suppliers
$7.00/5g
1282513-03-4
16 suppliers
inquiry
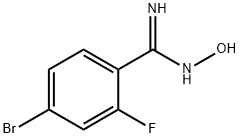
635702-31-7
16 suppliers
$78.29/1g
1282513-03-4
16 suppliers
inquiry
![9-bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine-2-carboxylicacid](/CAS/GIF/1282516-74-8.gif)
1282516-74-8
11 suppliers
inquiry
1282513-03-4
16 suppliers
inquiry