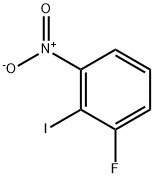
2-IODO-3-FLUORONITROBENZENE synthesis
- Product Name:2-IODO-3-FLUORONITROBENZENE
- CAS Number:122455-36-1
- Molecular formula:C6H3FINO2
- Molecular Weight:267
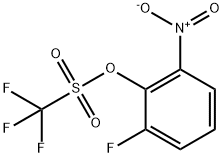
122455-35-0
2 suppliers
inquiry
122455-36-1
67 suppliers
$45.00/100mg
Yield: 81%
Reaction Conditions:
with lithium iodide in 1-methyl-pyrrolidin-2-one at 132;
Steps:
78
EXAMPLE 78 4-Amino-8-(2,4-dimethoxyphenyl)-7-fluoro-N-propylcinnoline-3-carboxamide Synthetic Scheme for Making Compound 78: A 2 L, 3-necked flask equipped with a mechanical stirrer was charged with 4-amino7-fluoro-8-iodo-N-propylcinnoline-3-carboxamide (40.5 g, 108.2 mmol), DME (700 mL, anhydrous), and ethanol (200 mL, absolute). A nitrogen dispersion tube was fitted into the suspension and the mixture was stirred until a solution was obtained. Water (300 mL) and PdCl2(PPh3)2 (7.6 g, 10 mol %) were added. After 5 minutes, 2,4-dimethoxyphenyl boronic acid (39.4 g, 216.5 mmol) was added followed by cesium carbonate (70.3 g, 216.5 mmol). Nitrogen was bubbled through the suspension for 5 minutes. The mixture was heated to approximately 80° C. Additional 7:3:2 DME:H2O:EtOH (340 mL) was added as the reflux started. The reaction was refluxed 18 hours and then cooled to room temperature, diluted with ethyl acetate (1.5 L), and washed with water (3×500 mL). The aqueous layers were extracted with ethyl acetate (3×150 mL). The combined organic layers were stirred for 1 hour with 40 g of DARCO, dried over sodium sulfate, and filtered through Celite. The solids were washed with 5% methanol in chloroform (3×200 mL) and the filtrates concentrated to a dark semisolid. This was taken up in 200 mL 1% methanol in chloroform and warmed to solubilize the material. The solution was divided into two portions. Each portion was filtered through Whatman fluted filter paper onto a 330 g silica gel column and eluted with 5% ethyl acetate in dichloromethane. (Note: Some solid catalyst appeared to be removed via the filter paper.) The purest fractions from each column were combined in 5-10% ethyl acetate in dichloromethane. The solution was concentrated to approximately 200 mL, diluted with hexane (200 mL), and let stand at room temperature overnight. The resulting solids were isolated by filtration, washed with ether (3 times), and dried under vacuum at room temperature to afford the desired product (26.4 g, 63%). 1H NMR (500.333 MHz, CDCl3) δ 8.51 (bs, 1H), 7.86 (dd, J=9.4, 5.2 Hz, 1H), 7.50 (t, J=8.8 Hz, 1H), 7.27 (d, J=9.2, 1H), 6.66 (dd, J=8.2, 2.3 Hz, 1H), 6.63 (d, J=2.3 Hz, 1H), 3.87 (s, 3H), 3.71 (s, 3H), 3.44 (q, J=6.7 Hz, 2H), 1.64 (sextet, J=7.3 Hz, 2H), 0.99 (t, J=7.4 Hz, 3H). MS APCI, m/z=385 (M+H). HPLC: 2.61 min.
References:
AstraZeneca AB US2007/142328, 2007, A1 Location in patent:Page/Page column 98-99
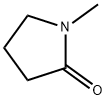
872-50-4
991 suppliers
$10.00/100ML
122455-35-0
2 suppliers
inquiry
122455-36-1
67 suppliers
$45.00/100mg

402-67-5
277 suppliers
$6.00/5g
122455-36-1
67 suppliers
$45.00/100mg