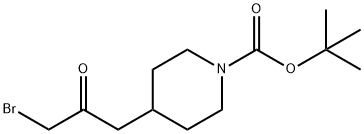
tert-butyl 4-(3-bromo-2-oxopropyl)piperidine-1-carboxylate synthesis
- Product Name:tert-butyl 4-(3-bromo-2-oxopropyl)piperidine-1-carboxylate
- CAS Number:473795-43-6
- Molecular formula:C13H22BrNO3
- Molecular Weight:320.22
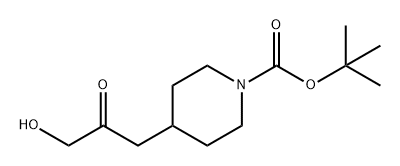
1580440-22-7
0 suppliers
inquiry
473795-43-6
9 suppliers
inquiry
Yield:473795-43-6 56%
Reaction Conditions:
with carbon tetrabromide;triphenylphosphine in dichloromethane; for 3 h;
Steps:
To a stirred and cooled (0 °C) solution of ethyl 2-hydroxyacetate (20.0 g, 19.0 mmol) in tetrahydrofuran (250 mL) was added tetrabutylammonium iodide (7.01 g, 19.0 mmol) and sodium hydride (60% in mineral oil, 7.60 g, 19.0 mmol). The mixture was stirred at 0 °C for 30 minutes before adding benzyl bromide (32.3 g, 19.0 mmol). The reaction was stirred overnight and allowed to warm to room temperature. The mixture was then cooled (0 °C), quenched with aqueous ammonium chloride (100 mL) and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried (Na2S04) and concentrated. The residue was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford ethyl 2- (benzyloxy)acetate as a yellow oil (14.7 g, 57%). To a stirred and cooled (0 °C) solution of this product (13.6 g, 70.0 mmol) in tetrahydrofuran (150 mL) was added dimethyl methylphosphonate (11.3 g, 91.1 mmol) followed by a 2.0 M solution of lithium diisopropylamide in tetrahydrofuran/heptane/ethylbenzene (74.0 mL, 148 mmol). The reaction was stirred at 0 °C for 3 hours before quenching with enough 5.0 M aqueous hydrochloric acid to bring the pH to ~4. The resulting mixture was then extracted with ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated. The residue was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford dimethyl (3-(benzyloxy)-2-oxopropyl)phosphonate as a light yellow oil (10.1 g, 54%). To a stirred and cooled (0 °C) solution of this intermediate (9.89 g, 36.4 mmol) in tetrahydrofuran (100 mL) was added sodium hydride (60% in mineral oil; 1.60 g, 40.0 mmol). The mixture was stirred at 0 °C for 30 minutes before adding, dropwise, a solution of tert-butyl 4-oxo-piperidine-l-carboxylate (5.79 g, 29.1 mmol) in tetrahydrofuran (50 mL). The resulting mixture was stirred at 0 °C for 2 hours, then quenched with saturated aqueous ammonium chloride and extracted with ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04), and concentrated. The residue was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford tert-butyl 4-(3-(benzyloxy)-2- oxopropylidene)piperidine-l-carboxylate as a light yellow oil (6.50 g, 52%). This material (6.50 g, 18.8 mmol), 10% Pd/C (1.00 g) and ethyl acetate (50 mL) were placed in a Parr bottle and hydrogenated for 5 hours at room temperature. The mixture was filtered through Celite and concentrated to afford tert-butyl 4-(3-hydroxy-2- oxopropyl)piperidine-l-carboxylate as a yellow oil (4.80 g, 99%). To a stirred solution of this product (4.80 g, 18.7 mmol) in methylene chloride (8 mL) was added carbon tetrabromide (12.4 g, 37.4 mmol) and triphenylphosphine (9.80 g, 37.4 mmol). After 3 hours, the reaction was concentrated and the residue purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford tert-butyl 4-(3-bromo-2- oxopropyl)piperidine-l-carboxylate as a white solid (3.30 g, 56%). To a stirred and cooled (0 °C) solution of this product (3.30 g, 10.3 mmol) in methylene chloride (50 mL) was added trifluoroacetic acid (12.0 mL, 145 mmol). The mixture was stirred at 0 °C for 30 min before concentrating to afford crude l-bromo-3-(piperidin-4-yl)acetone trifluoroacetate, which was used without purification in the next step. To a stirred solution of diisopropylethylamine (20 mL) in acetonitrile (800 mL) at reflux was added a solution of the crude intermediate in acetonitrile (150 mL), dropwise over 4 hours (syringe pump). Reflux was continued overnight and then the mixture was concentrated. The resulting residue was partitioned between aqueous potassium carbonate solution and 9: 1 (v/v) chloroform/methanol. The organic layer was combined with a second extract using the same solvent mixture, dried (Na2S04) and concentrated. The crude material was purified flash chromatography over silica using a 95/4.5/0.5 (v/v) chloroform/methanol/ammonium hydroxide eluant to afford l-azabicyclo[3.2.2]nonan-3- one as a brown solid (770 mg, 54%). To a stirred and cooled (0 °C) solution of this intermediate (770 mg, 5.54 mmol) in tetrahydrofuran (10 mL), was added lithium aluminum hydride (211 mg, 5.54 mmol), portion wise. After vigorous hydrogen gas evolution ceased, the reaction mixture was allowed to warm to room temperature and then heated at reflux for 1 hour. The solution was cooled to 0 °C and quenched by the successive dropwise addition of water (0.2 mL), 10% aqueous sodium hydroxide solution (0.2 mL) and water again (0.6 mL). The colorless precipitate was removed by filtration through Celite, which was subsequently washed with tetrahydrofuran. The combined filtrate was dried (Na2S04) and concentrated to afford the title compound as yellow solid (770 mg, 99%).
References:
WO2014/43068,2014,A1 Location in patent:Page/Page column 167-169
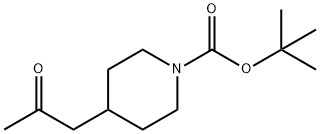
206989-54-0
42 suppliers
$333.00/1g
473795-43-6
9 suppliers
inquiry
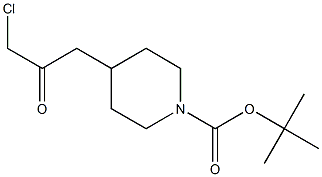
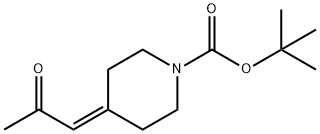
473795-40-3
2 suppliers
inquiry
473795-43-6
9 suppliers
inquiry
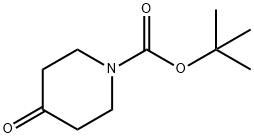
79099-07-3
543 suppliers
$5.00/5g
473795-43-6
9 suppliers
inquiry