
5-BroMo-2-Methoxy-4-Methyl-benzoic acid synthesis
- Product Name:5-BroMo-2-Methoxy-4-Methyl-benzoic acid
- CAS Number:90326-61-7
- Molecular formula:C9H9BrO3
- Molecular Weight:245.07
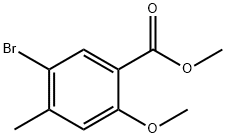
39503-58-7
32 suppliers
$48.00/100mg
90326-61-7
25 suppliers
inquiry
Yield:90326-61-7 82%
Reaction Conditions:
Stage #1: methyl 5-bromo-2-methoxy-4-methylbenzoatewith sodium hydroxide in ethanol;water; for 0.5 h;Heating / reflux;
Stage #2: with hydrogenchloride in water; pH=2;
Steps:
12
A 250 ml 3-neck RBF fitted with a magnetic stirring bar, a thermometer and adropping funnel was charged with methyl 2-methoxy-4-methylbenzoate 10b (10 g,0.056 mol) and AcOH (70 ml). To this solution bromine (3 ml, 0.06 mol) was addeddrop-wise while maintaining the reaction temperature below 25°C. When bromineaddition was complete, the reaction mixture was stirred at RT for 2 hours, thenpoured into 600 ml_ of cold water and the pH was adjusted to 8-9 with Na2CO3. Thecrude product was extracted with Et2O (3 X 300 ml), the combined extracts washedwith brine, dried over sodium sulfate and the solvent was removed under vacuum togive methyl 5-bromo-2-methoxy-4-methylbenzoate as an orange oil, which solidifiedon standing (14.3 g, 99% yield, >90% purity by 1H NMR).Methyl 5-bromo-2-methoxy-4-methylbenzoate (11.64 g, 0.045 mol) was dissolved ina mixture of EtOH:H2O (1:1 ratio, 150 mL), NaOH (5.8 g, 0.145 mol) was added andreaction mixture was refiuxed for 30 min. The EtOH solvent was removed from themixture by distillation which leads to precipitation of the desired product as itssodium salt. The residue was diluted with H2O (75 mL) and the pH was adjusted to~2 with concentrated HCI. The precipitate was collected by filtration, washed withH2O (3 X 50 mL) and air dried to give pure 5-bromo-2-methoxy-4-methylbenzoicacid 12a (9 g, 82% yield) as a beige solid. A mixture of 5-bromo-2-methoxy-4-methylbenzoic acid 12a (9 g, 0.037 mol), thionylchloride (5 ml, 0.069 mol) and benzene (90 mL) was heated to reflux for 4 hours, then evaporated to dryness at reduced pressure. The residue of thechloroanhydride intermediate was dissolved in dry benzene (40 ml) and pouredslowly into an ice cooled flask containing 150 ml of concentrated ammoniumhydroxide solution. The reaction mixture was then stirred for 1 hour at RT and thesolid material which precipitated was collected by filtration, washed with water (3 X25 ml), benzene (2 X 20 ml_) and air dried to afford pure 5~bromo-2-methoxy-4-methylbenzamide 12b (8.5 g, 95% yield) as beige solid.A 250 ml 3-neck RBF fitted with a magnetic stirring bar, a thermometer, a droppingfunnel and a condenser was charged with 5-bromo-2-methoxy-4-methylbenzamide12b (6 g, 24.6 mmol), anhydrous MeOH (40 ml) and 25% NaOMe solution in MeOH(21 ml, 98.4 mmol). This slurry was cooled to 5°C and bromine (1.4 ml, 27.1mmol) was added drop wise causing ah exothermic reaction and the formation of acloudy solution. The reaction mixture was stirred for 30 min at RT, and then heatedto reflux for 1 hour. The solvent was removed under vacuum, the residue was re-dissolved in EtOH (50 mL) and KOH (5.5 g, 98.4 mmol) was added. The mixture. was allowed to react under reflux for 16 hours in order to hydrolyze the intermediatemethylcarbamate. Then the reaction mixture was diluted with H2O (300 mL) and theproduct was extracted into Et2O (3 X 100 ml). The combined organic layers werefiltered through a pad of silica gel and evaporated to dryness to give 5-bromo-2-methoxy-4-methylaniline 12c (4.8 g, 90% yield) pure by 1H NMR as a light brownsolid.A suspension of 5-bromo-2-methoxy-4-methylaniline hydrochloride [formed from thereaction of 5-bromo-2~methoxy-4-methylaniline (4.32 g, 20 mmol) with cone. HCI (20ml)] was cooled to 0°C and an aqueous solution of NaNO2 (1.224 g, 24 mmol in 5mL H2O) was added slowly, keeping the reaction temperature below 5°C. Whenaddition of the nitrite was complete, the reaction mixture was stirred for 30 min at0°C, then poured into a previously prepared mixture containing 30% SO2 in AcOH(30 mL), a solution of CuCI22H2O (5.13 g, 30 mmol) in 15 mL H2O and benzene (20mL). The reaction mixture was heated slowly to 35°C, at which point the evolutionof nitrogen gas was observed. When gas evolution had stopped, the reactionmixture was diluted with water (200 mL), the organic layer was separated, washedwith an aqueous solution of saturated NaHCO3, dried over Na2SO4 and evaporated to dryness to give the crude sulfonylchloride 12d as a brown oil (~3 g). Thisintermediate was dissolved in hexane (20 ml_), a solution of concentrated NH4OH(20 mL) was added and reaction mixture was stirred vigorously at RT overnight.The brown solid formed was collected by filtration and treated with 10% KOHsolution (20 ml). Any insoluble impurities were removed by filtration and the filtratewas acidified to give a beige precipitate which was filtered, washed with water andair dried to give the pure product 5-bromo-2-methoxy-4-methylbenzenesulfonamide12e (0.586 g, 10.4 % overall yield).A dry 25 ml 3-neck RBF, fitted with a magnetic stirring bar, a thermometer andseptum with Ar inlet was charged with 5-bromo-2-methoxy-4-methylbenzene-sulfonamide 12e (0.586 g, 2.1 mmol), anhydrous THF (5 mL) and HMPA (0.37 mL,2.1 mmol). The mixture was cooled to -75°C and a 2.0 M solution of LDA (4.2 mL,8.4 mmol) was added drop wise via a syringe. The reaction mixture was stirred at -75°C for 1.5 hours and then cannulated into a flask containing 20 g of dry ice and 5mL THF and left to warm up slowly to RT. The solvent was removed under reducedpressure and H2O (20 mL) was added to the residue. At this point, some of theunreacted starting material precipitated and was removed by filtration. The filtratewas acidified and the solution was extracted with EtOAc (3 X 20 mL). Thecombined organic layers were evaporated to dryness, the residue was treated withan aqueous solution of saturated NaHCO3 (10 mL) and the insoluble material wasseparated by filtration. The filtrate was acidified with concentrated HCI and thecloudy solution extracted again with EtOAc (3X5 mL). The combined organiclayers were concentrated to ~1 mL, causing crystallization of the product, which wascollected by filtration to give pure (2-bromo-5-methoxy-4-sulfamoylphenyl)acetic acid(20mg)12f(~3%yield).
References:
WO2004/108673,2004,A2 Location in patent:Page 47-49
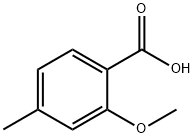
704-45-0
132 suppliers
$14.00/1g
90326-61-7
25 suppliers
inquiry

50-85-1
346 suppliers
$10.00/5g
90326-61-7
25 suppliers
inquiry

81245-24-1
125 suppliers
$42.00/1g
90326-61-7
25 suppliers
inquiry
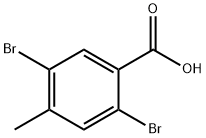
20871-01-6
15 suppliers
$332.00/250mg
90326-61-7
25 suppliers
inquiry