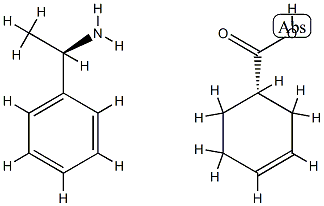
3-Cyclohexene-1-carboxylic acid, (1S)-, compd. with (αR)-α-methylbenzenemethanamine (1:1) synthesis
- Product Name:3-Cyclohexene-1-carboxylic acid, (1S)-, compd. with (αR)-α-methylbenzenemethanamine (1:1)
- CAS Number:67976-82-3
- Molecular formula:C15H21NO2
- Molecular Weight:247.3327
![(1S,4S,5S)-4-Bromo-6-oxabicyclo[3.2.1]octan-7-one](/CAS/20200515/GIF/139893-81-5.gif)
Yield:67976-82-3 240 g
Reaction Conditions:
in water;acetone at 10 - 50; for 23 h;Solvent;Temperature;
Steps:
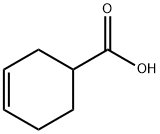
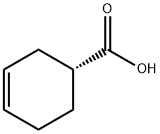
1 (S)-3-Cyclohexene-1-carboxylic acid (R)-α-phenylethylamine salt (2-b)
Reference Example 1
(S)-3-Cyclohexene-1-carboxylic acid (R)-α-phenylethylamine salt (2-b)
3-Cyclohexene-1-carboxylic acid (1) (1.0 kg) was dissolved in 4.8% aqueous acetone (7.5 l), and, a solution (500 ml) prepared by dissolving (R)-α-phenylethylamine (3) (624.3 g) in 4.8% aqueous acetone was gradually added to the above obtained solution at 50° C.
The obtained mixture was stirred at the same temperature as described above for 4 hours.
The suspension was cooled to 35° C., and it was stirred at the same temperature as described above for 16 hours.
Then, the suspension was further stirred at 10° C. for 3 hours.
The suspension was filtrated under reduced pressure, so that 837.1 g of the title salt compound was obtained in the form of a crystal.
The optical purity thereof was 63% de.
Subsequently, 4.8% aqueous acetone (5.6 l) was added to 700 g of the obtained salt compound, and the obtained mixture was stirred under heating to reflux for 5 hours, and at 30° C. for 13 hours.
After that, the reaction mixture was stirred under cooling on ice for 3 hours.
The suspension was filtrated under reduced pressure to obtain 519.4 g of the title salt compound in the form of a crystal.
The optical purity was 81% de.
Further, 4.8% aqueous acetone (4.0 l) was added to 500 g of the obtained salt compound, and the obtained mixture was stirred under heating to reflux for 5 hours, and at 30° C. for 13 hours, and after that, the reaction mixture was stirred at 10° C. for 3 hours.
The suspension was filtrated under reduced pressure to obtain 398.5 g of the title salt compound in the form of a crystal.
The optical purity thereof was 91% de.
Finally, 4.8% aqueous acetone (2.4 l) was added to 300 g of the obtained salt compound, and the obtained mixture was stirred under heating to reflux for 5 hours, and at 30° C. for 13 hours, and after that, the reaction mixture was stirred at 10° C. for 3 hours.
The suspension was filtrated under reduced pressure to obtain 240.0 g of the title compound (2-b) in the form of a crystal.
The optical purity thereof was 97% de.
1H-NMR (D2O) δ: 1.50-1.63 (1H, m), 1.66 (3H, d, J=6.9 hz), 1.86-1.95 (1H, m), 1.98-2.25 (4H, m), 2.32-2.43 (1H, m), 4.56 (1H, q, J=6.9 Hz), 5.70-5.80 (2H, m), 7.40-7.55 (5H, m).
References:
US2015/353577,2015,A1 Location in patent:Paragraph 0085; 0086
5709-98-8
179 suppliers
$13.00/1g
67976-82-3
9 suppliers
inquiry

6493-77-2
184 suppliers
$16.00/1g
67976-82-3
9 suppliers
inquiry
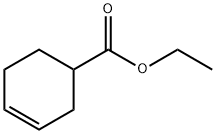
15111-56-5
42 suppliers
$24.73/1gm:
67976-82-3
9 suppliers
inquiry