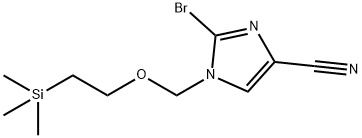
1-((2-(Trimethylsilyl)Ethoxy)Methyl)-2-Bromo-1H-Imidazole-4-Carbonitrile synthesis
- Product Name:1-((2-(Trimethylsilyl)Ethoxy)Methyl)-2-Bromo-1H-Imidazole-4-Carbonitrile
- CAS Number:854044-51-2
- Molecular formula:C10H16BrN3OSi
- Molecular Weight:302.24
Yield:854044-51-2 800 mg
Reaction Conditions:
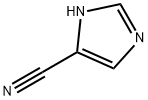
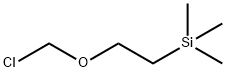
Stage #1: (2-trimethylethylsilylethoxy)methyl chloride;1H-imidazole-4-carbonitrilewith potassium carbonate in acetone at 10 - 20;
Stage #2: with N-Bromosuccinimide in tetrachloromethane at 60 - 75;
Steps:
148.1; 148.2
To a suspension of lH-imidazole-4-carbonitrile (1 g, 10.76 mmol) and potassium carbonate (2.96 g, 21.5 mmol) in acetone (20 mL) at 10 °C, was added SEMCl (1.98 g, 11.8 mmol) dropwise over 30 min, while the internal temperature was kept below 15 °C. The reaction was then allowed to warm to rt and stirred overnight. After the reaction was completed, the reaction was quenched by cold water (50 mL) and extracted with EA (50 mL x 3). The combined organic layer was dried over sodium sulfate (30 g), filtered and concentrated. The concentrated residue was purified by silica gel chromatography (eluent: EA/n-Hex = 1/4) to afford 1.3 g of a mixture of the title compound and its regioisomer 2a. LC-MS (Method A) (ESI+): m/z 224 (M+H)+. Step 2: Synthesis of 2-bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-lH- imidazole-4- carbonitrile: A solution of 1 -((2-(trimethylsilyl)ethoxy)methyl)-l H-imidazole-4-carbonitrile 2 and l-((2-(trimethyl silyl)ethoxy)methyl)-lH-imidazole-5-carbonitrile 2a (1.30 g, 5.83 mmol) in CCLi (30 mL) at 60 °C was added NBS (1.14 g, 6.41 mmol) portion-wise over 5 min. An exothermic reaction was observed, and the internal temperature increased to 75 °C. After addition, the reaction was stirred at 60 °C overnight. The reaction was allowed to cool to rt, and the resulting suspension was filtered. The filter cake was washed with CCU (20 mL). The combined filtrate was washed with a saturated NaHCCb solution (60 mL). After separation, the organic layer was dried over sodium sulfate (30 g), filtered and concentrated. The residue was purified by silica gel chromatography (eluent: EA/n-Hex = 1/10) to afford 800 mg of the title compound. LC-MS (Method A) (ESI+): m/z 324 (M+Na)+; lH-NMR (300 MHz, CDCh) <5 7.63 (s, 1H), 5.30 (s, 2H), 3.55 (t, J = 8.1 Hz, 2H), 0.93 (t , J= 8.1 Hz, 2H), 0.05 (s, 9H).
References:
WO2020/132269,2020,A1 Location in patent:Paragraph 0692-0693
![1-{[2-(trimethylsilyl)ethoxy]methyl}-1H-imidazole-4-carbonitrile](/CAS/20180703/GIF/854044-50-1.gif)
854044-50-1
4 suppliers
inquiry
854044-51-2
34 suppliers
inquiry
![1H-Imidazole-5-carbonitrile, 1-[[2-(trimethylsilyl)ethoxy]methyl]-](/CAS/20210305/GIF/885693-61-8.gif)
76513-69-4
358 suppliers
$6.00/1g
854044-51-2
34 suppliers
inquiry
![1H-Imidazole-4-carboxylic acid, 2-bromo-1-[[2-(trimethylsilyl)ethoxy]methyl]-](/CAS/20210305/GIF/1403334-53-1.gif)
1403334-53-1
2 suppliers
inquiry
854044-51-2
34 suppliers
inquiry