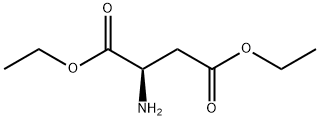
D-Aspartic acid, 1,4-diethyl ester synthesis
- Product Name:D-Aspartic acid, 1,4-diethyl ester
- CAS Number:20268-79-5
- Molecular formula:C8H15NO4
- Molecular Weight:189.21
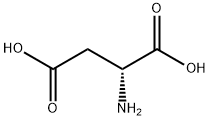
1783-96-6
566 suppliers
$8.00/25g
20268-79-5
0 suppliers
inquiry
Yield:20268-79-5 99%
Reaction Conditions:
Stage #1: acetyl chloride in ethanol at 0 - 20; for 0.5 h;
Stage #2: (2R)-aspartic acid in ethanol; for 2 h;Heating / reflux;
Steps:
80
Acetyl chloride (54.6 mL, 0.75 mol) was added drop-wise into ethanol (316 mL) at 0-5 C. When the addition was completed, the ice bath was removed and the solution was allowed to stir while warming to room temperature for another 30 mins. D-aspartic acid 1 (25 g, 0.188 mol) was then added. The reaction mixture was refluxed for 2 hours. The reaction solution was then concentrated in vacuo and placed under high vacuum (0.4 mm Hg) overnight. Compound 2 was obtained as a white solid (42g, 99%) and used directly in the next step. [00469] (BOC)20 (44.7 g, 0.21mol) was added portion-wise over 10 mins to a 0 C solution of compound 1 (42 g, 0.19 mol), trimethyl amine (51.9 ml, 0.37 mol), dioxane (140 mL) and water (56 mL). After another 10 min, the ice bath was removed and the reaction mixture was stirred while warming to room temperature for another 2 hours. The reaction mixture was diluted in ethyl acetate (150 mL) and washed with 0.5 N HCI (200 mL x 3). The organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated in vacuo giving compound 3 (52 g, yield 97%) which was used directly in the next step. [00470] NaBH4 (54.4 g, 1.44 mol) was added portion-wise over 30 mins to a 0 C solution of compound 3 (52 g, 86.4 mmol) and ethanol (600 mL). The reaction mixture was extremely exothermic and great care was exercised during the addition of reducing agent. After the addition was complete, the reaction mixture was heated to reflux for 1 hour. The solution was cooled to ambient temperature and the reaction mixture solidified. The solid was broken-up to a slurry, which was then poured into brine (250 mL). The resulting mixture was filtered and the filtrate was concentrated in vacuo. The resulting residue was vigorously stirred with ether (200 mL x 5). The ether layers were successively decanted from the residue. The combined ether extracts were dried over magnesium sulfate, filtered, and the filtrate was concentrated in vacuo giving compound 4 as white solid (25.2g, yield 68%). [Note: Yield was 89% when performed on a 25 g (compound 3) scale,] [00471] t-Butyldiphenylchlorosilane (31.9 mL, 0.123 mol) was added to a solution of compound 4 (25.2 g, 0.123 moll, diisopropylethylamine (42.8 mL, 0.245 mol), and CH2Cl2 (500 mL). The reaction solution was stirred at ambient temperature for 24 hrs. The reaction solution was then washed with 0.5 N HCI (150 mL x 3) and brine (150 mL). The organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by flash chromatography (silica gel, 4:1 hexanes:EtOAc) to give compound 5 (42g, yield 77%). [Note: Yield was 85% when performed on 15 g (compound 4) scale.] [00472] Iodine (24 g, 94.7 mmol) was added portion-wise over 15 mins to a 0 C solution of compound 5 (28 g, 63.1mmol), Ph3P (24.8 g, 94.7 mmol), imidazole (6.4g, 94.7 mmol), diethyl ether (450 mL) and acetonitrile (150mL). The ice bath was removed and the reaction solution was allowed to wann to ambient temperature over 30 mins. The reaction was judged complete by TLC analysis (4:1 hexanes:EtOAc), The reaction was quenched with water (400 mL). The layers were separated and the aqueous layer was extracted by diethyl ether (100 mL). The combined organic layers were washed with saturated aqueous Na2SO3 (100 x 2) and brine (100 mL). The organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by flash column chromatography (silica gel, 4:1 hexanes:EtOAc) to give compound 6 (32 g, 92%).
References:
WO2005/107762,2005,A2 Location in patent:Page/Page column 266-268

112018-26-5
55 suppliers
$6.00/1g
20268-79-5
0 suppliers
inquiry

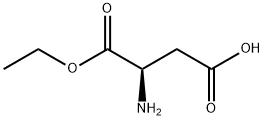
565461-05-4
1 suppliers
inquiry
20268-79-5
0 suppliers
inquiry
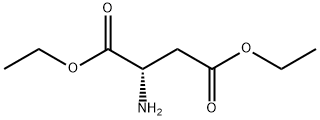
43101-48-0
15 suppliers
inquiry
20268-79-5
0 suppliers
inquiry