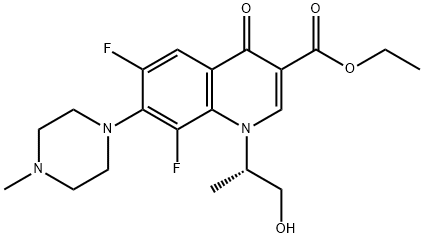
Levofloxacin Impurity 22 synthesis
- Product Name:Levofloxacin Impurity 22
- CAS Number:177472-29-6
- Molecular formula:C20H25F2N3O4
- Molecular Weight:409.43

109-01-3
675 suppliers
$5.00/5G
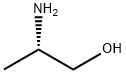
2749-11-3
418 suppliers
$14.56/5G
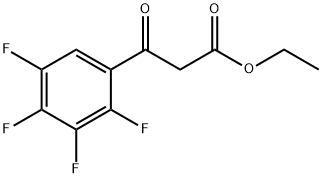
94695-50-8
198 suppliers
$17.00/5g
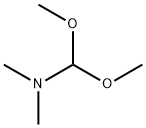
4637-24-5
646 suppliers
$5.00/25g
177472-29-6
10 suppliers
inquiry
Yield:177472-29-6 63.5%
Reaction Conditions:
Product distribution / selectivity;
Steps:
6
EXAMPLE 6; (S)-ethyl 6,8-difluoro-1-(1-hydroxypropan-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (compound of formula (VI) where R1 is COOEt) Alternatively, the title compound could be obtained in a one pot process starting from 3-oxo-3-(2,3,4,5-tetrafluorophenyl)propanoate: To a solution of 62 g (0.235 mol) of 3-oxo-3-(2,3,4,5-tetrafluorophenyl)propanoate in 450 mL of toluene, 68 mL (0.61 mol) of 1-methylpiperazine were added and the mixture heated at 70 °C for 80 minutes. The solution was cooled to room temperature and washed twice with a solution of water with sodium bicarbonate. The organic phase was dried by azeotropic distillation of toluene and the initial volume was recovered with more toluene. Dimethyl acetal of N,N-dimethylformamide 40 mL (0.30 mol) were added and the resultant mixture was heated at reflux for 2 and a half hours. The crude reaction mixture was cooled and washed twice with a solution of sodium bicarbonate, the organic phase was dried by azeotropic distillation and the original volume was recovered with more toluene. 19 mL of L- alaninol (0.24 mol) were added to the cooled solution at 15-20 °C and after stirring the reaction for 1 and a half hours, the mixture was washed twice with a solution of sodium bicarbonate, the organic phase was dried by azeotropic distillation and the original volume was recovered with more toluene. 56 mL (0.40 mol) of triethylamine were added and the mixture was cooled to 0-5 °C then 31 mL (0.43 mol) of acetyl chloride were added over 30 minutes. The reaction was left at 5-10 °C for 1 and a half hours, the crude reaction mixture was washed twice with a solution of sodium bicarbonate, the organic phase was dried by azeotropic distillation and the original volume was recovered with more toluene. 120 g (0.87 mol) of potassium carbonate and 1 g (0.003 mol) of tetrabuthylammonium bromide were added and the mixture was heated at reflux for three hours. The reaction was cooled to room temperature, filtered and the solid was washed with more toluene. The toluene was distilled under vacuum and stripped with the addition of methanol. 250 mL of methanol and 1 g (0.007 mol) of potassium carbonate were added and the mixture was left stirring for 1 hour. The potassium carbonate was neutralised with 1 mL of acetic acid and the solvent was evaporated under vacuum and stripped with ethyl acetate until a precipitate was formed. 300 mL of ethyl acetate were added and after stirring for 1 hour, the suspension was cooled at 5-10 °C for 2 hours then the solid was filtered and washed with cold ethyl acetate. 53 g of (S)-ethyl 6,8-difluoro-1-(1-hydroxypropan-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate were obtained, and a second crop of 7 g was recovered after concentrating the filtrate to give a total of 61 g (63.5 % yield for the six steps).
References:
EP1939206,2008,A1 Location in patent:Page/Page column 10-11