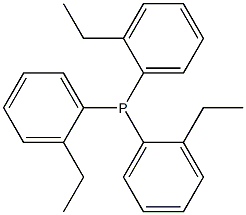
Phosphine, tris(2-ethylphenyl)- synthesis
- Product Name:Phosphine, tris(2-ethylphenyl)-
- CAS Number:50777-27-0
- Molecular formula:C24H27P
- Molecular Weight:346.4449
1973-22-4
210 suppliers
$10.00/5g
50777-27-0
1 suppliers
inquiry
Yield:50777-27-0 67%
Reaction Conditions:
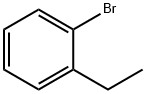
Stage #1: 1-bromo-2-ethylbenzenewith n-butyllithium in tetrahydrofuran;hexane at -60; for 2 h;Inert atmosphere;
Stage #2: with phosphorus trichloride in tetrahydrofuran;hexane at -54 - 20;Inert atmosphere;
Steps:
2.E Tris(2-ethylphenyl)phosphine L3
An oven dried, 50 mL 3-neck round bottom flask equipped with a stir bar was fitted with a thermometer adapter, a nitrogen inlet, and a rubber septum. The apparatus was vac-filled three times with nitrogen and charged with 40 1-bromo-2-ethylbenzene (0.55 mL, 740 mg, 4.0 mmol, 1.0 equiv.) and 41 THF (6.5 mL, 1.6 Molar). The RBF was lowered into a dry ice/acetone bath and allowed to equilibrate for over 20 minutes. nbutyllithium (1.6 Molar in hexanes, 2.4 mL, 3.8 mmol, 0.95 equiv.) was added dropwise over 10 minutes, keeping temperature below -60° C. The reaction appearance changed from colorless to green. Following the addition, the reaction mixture was allowed to stir for one hour and 50 minutes in the dry ice/acetone bath. (0137) An oven dried 40 mL vial equipped with a stir bar was backfilled with nitrogen and charged with 42 phosphorus trichloride (0.36 mL, 0.565 g, 4.12 mmol) and THF (8.0 mL; 0.515 Molar). 2.1 mL of the resulting 43 PCl3 solution (1.08 mmol, 0.27 eq) was transferred to the reaction mixture over 10 minutes (maintaining a temperature below -54° C.). Following the addition, the reaction mixture was allowed to stir and warm to room temp overnight. The reaction mixture was cooled to 0° C. in an ice/water bath and was quenched with 0.7 mL water and 8 mL NH4Cl following equilibration. The quenched solution was then transferred to a separatory funnel, rinsing with water and toluene. After removing the organic layer, the aqueous layer was extracted with toluene (2×10 mL). Combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated by rotary evaporation to afford a sticky white/yellow solid was allowed to dry overnight on high vac. The crude product was purified by normal phase column chromatography (4.5×9 cm silica gel column, isocratic 100% hexanes), affording the pure 45 L3 as a white solid (251.4 mg, 0.7256 mmol, 67% yield). (0138) 1H NMR (500 MHz, acetone-d6) δ 7.30-7.23 (m, 6H), 7.05 (m, 3H), 6.70 (m, 3H), 2.76 (dq, J=7.5, 1.2 Hz, 6H), 1.07 (t, J=7.5 Hz, 9H). 13C NMR (126 MHz, acetone-d6) δ 149.31 (d, JC-P=25.7 Hz), 135.60 (d, JC-P=11.6 Hz), 134.60, 130.06, 129.45 (d, JC-P=5.0 Hz), 126.96, 28.19 (d, JC-P=22.4 Hz), 15.69 (d, JC-P=3.0 Hz). 31P NMR (202 MHz, acetone-d6, referenced to H3PO4 in D2O) δ -35.62. Rf=0.31 on normal phase TLC in 100% hexanes. HRMS (ES+) Calculated for C24H28P: 347.1929; Found: 347.1926.
References:
US2018/305381,2018,A1 Location in patent:Paragraph 0112; 0136-0138