| Identification | More | [Name]
Voriconazole | [CAS]
137234-62-9 | [Synonyms]
2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1h-1,2,4-triazol-1-yl)butan-2-ol
VORICONAZOLE
(2R,3S/2S,3R)-3-(5-FLUORO-4-PYRIMIDINYL)-2-(2,4-DIFLUOROPHENYL)-1-(1H-1,2,4-TRIA
(aR,bS)-a-(2,4-Difluorophenyl)-5-fluoro-b-methyl-a-(1H-1,2,4-triazol-1-ylmethyl)-4-pyrimidineethanol
UK-10949
Vorionazole
(2R,3S)-2-(2,4-DIFLUOROPHENYL)-3-(5-FLUOROPYRIMIDIN-4-YL)-1-(1H-1,2,4-TRIAZOL-1-YL)BUTAN-2-OL
(aR,S)-a-(2,4-Difluorophenyl)-5-fluoro--methyl-a-(1H-1,2,4-triazol-1-ylmethyl)-4-pyrimidineethanol
UK-109496 | [EINECS(EC#)]
629-701-5 | [Molecular Formula]
C16H14F3N5O | [MDL Number]
MFCD00905717 | [Molecular Weight]
349.31 | [MOL File]
137234-62-9.mol |
| Chemical Properties | Back Directory | [Appearance]
Cyrstalline Solid | [Melting point ]
127-130°C | [alpha ]
D25 -62° (c = 1 in methanol) | [Boiling point ]
508.6±60.0 °C(Predicted) | [density ]
1.42±0.1 g/cm3(Predicted) | [Fp ]
9℃ | [storage temp. ]
2-8°C | [solubility ]
DMSO: >20mg/mL | [form ]
white powder | [pka]
11.54±0.29(Predicted) | [color ]
White to Almost white | [Usage]
An antifungal. An Ergosterol Biosynthesis inhibitor | [Merck ]
14,10033 | [InChI]
InChI=1/C16H14F3N5O/c1-10(15-14(19)5-20-7-22-15)16(25,6-24-9-21-8-23-24)12-3-2-11(17)4-13(12)18/h2-5,7-10,25H,6H2,1H3/t10-,16+/s3 | [InChIKey]
BCEHBSKCWLPMDN-MGPLVRAMSA-N | [SMILES]
[C@@](C1C=CC(F)=CC=1F)(O)(CN1N=CN=C1)[C@H](C1=NC=NC=C1F)C |&1:0,16,r| | [CAS DataBase Reference]
137234-62-9(CAS DataBase Reference) |
| Safety Data | Back Directory | [Hazard Codes ]
Xn | [Risk Statements ]
R22:Harmful if swallowed.
R36/38:Irritating to eyes and skin . | [Safety Statements ]
S26:In case of contact with eyes, rinse immediately with plenty of water and seek medical advice .
S36:Wear suitable protective clothing . | [RIDADR ]
UN1230 - class 3 - PG 2 - Methanol, solution | [WGK Germany ]
3 | [RTECS ]
UV9145000 | [HazardClass ]
6.1 | [PackingGroup ]
III | [HS Code ]
29335900 |
| Hazard Information | Back Directory | [Description]
Voriconazole was introduced in the US for the treatment of acute invasive aspergillosis,
candidosis and other emerging fungal infections seen in immuno compromised patients. It
can be synthesized in 3 steps by reaction of readily available 6-( 1 -bromoethyl)-4-chloro-5
fluoropyrimidine with I-(2,4-difluorophenyl)-2-(1,2,4-triazol-I-yl) ethanone in the presence
of zinc metal. The resulting racemic mixture was submitted to a reductive dechlorination
step followed by resolution with (R)-camphorsulfonic acid. Voriconazole is structurally
related to fluconazole (Pfizer, diflucan?) and acts by inhibiting the cytochrome P450-
dependant enzyme 14a-sterol demethylase of ergosterol synthesis (thereby resulting in
the formation of a cell membrane with abnormal characteristics and accumulation of toxic
sterol intermediates). Voriconazole was more active than itraconazole and fluconazole
against Cryptococcus neoformans and a variety of Candidas species such as C. albicans,
C. glabrata C. krusei. It also exhibits similar or superior activity compared to amphotericin
B and itraconazole against filamentous fungi such as Aspergillus, an important pathogen
which is not susceptible to fluconazole. In clinical trials, voriconazole was effective in the
treatment of neutropenic patients with acute invasive aspergillosis, non-neutropenic
patients with chronic invasive aspergillosis and HIV patients with oropharyngeal
candidiasis. Voriconazole is available as oral or intravenous formulations. Following oral
administration, absorption is rapid and the bioavailability is greater than 80%. Voriconazole
exhibits non linear pharmacokinetics, a large volume of distribution (2 L/Kg) and a
relatively short half-life (6 h). It was extensively metabolized via hepatic cytochrome P450
and has a drug interactions potential similar to itraconazole. Voriconazole was generally
well tolerated, the most common treatment-related adverse events were transient visual
disturbances. | [Chemical Properties]
Cyrstalline Solid | [Originator]
Pfizer (UK) | [Uses]
Voriconazole is an antifungal (systemic) that belong to an ergosterol biosynthesis inhibitor. It is used to treat serious fungal or yeast infections, such as aspergillosis (fungal infection in the lungs), candidemia (fungal infection in the blood), esophageal candidiasis (candida esophagitis), or other fungal infections (infections in the skin, stomach, kidney, bladder, or wounds). | [Definition]
ChEBI: A triazole-based antifungal agent used for the treatment of esophageal candidiasis, invasive pulmonary aspergillosis, and serious fungal infections caused by Scedosporium apiospermum and Fusarium spp. It is an inhibitor of cytochr
me P450 2C9 (CYP2C9) and CYP3A4. | [Indications]
Voriconazole (Vfend), a derivative of fluconazole, is a
second-generation triazole that has improved antifungal
activity against Aspergillus and Fusarium spp., P.
boydii, Penicillium marneffei, and fluconazole-resistant
Candida spp. Like fluconazole, voriconazole has high
oral bioavailability and good cerebrospinal fluid penetration,
but unlike fluconazole, it undergoes extensive
hepatic metabolism and is highly protein bound. No significant
amount of bioactive drug is excreted into the
urine. Dosage reduction is necessary with severe hepatic
insufficiency but not with renal insufficiency. | [Manufacturing Process]
A solution of 3-(4-chloro-5-fluoropyrimidin-6-yl)-2-(2,4-difluorophenyl)-1-(1H-
1,2,4-triazol-1-yl)butan-2-ol, enantiomeric pair B (0.307 g, 0.8 mmol) in
ethanol (20 ml) was hydrogenated at atmospheric pressure and at room
temperature in the presence of 10% palladium-on-charcoal (30 mg) and
sodium acetate (0.082 g, 1 mmol). After 5 hours a further 10 mg of 10%
palladium-on-charcoal was added and hydrogenation was continued for an
additional 1 hour. The catalyst was removed by filtration and the filtrate was
concentrated in vacuo. 'Flash' chromatography of the residue on silica using
97:3 ethyl acetate/methanol as the eluent provided, after combination and
evaporation of appropriate fractions and trituration with diethyl ether, the 2-
(2,4-difluorophenyI)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-I-yl)butan-
2-ol enantiomeric pair B, (0.249 g, 89%), m.p. 127°C.
2-(2,4-DifluorophenyI)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-
yl)butan-2-ol enantiomeric pair A was prepared by a similar method using 3-
(4-chloro-5-fluoropyrimidin-6-yl)-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-
yl)butan-2-ol, enantiomeric pair A as a starting material. This gave the
product with m.p. 137°C. | [Brand name]
Vfend (Pfizer). | [Therapeutic Function]
Antifungal | [Antimicrobial activity]
The spectrum includes most fungi that cause human disease:
dimorphic fungi (Blast. dermatitidis, Coccidioides spp., Hist. capsulatum,
Paracocc. brasiliensis, Pen. marneffei and Spor. schenckii),
molds (Aspergillus spp., Fusarium spp. and Scedosporium spp.),
dematiaceous fungi and yeasts (Candida spp., Cryptococcus
spp. and Trichosporon spp.). | [Acquired resistance]
Some fluconazole- and itraconazole-resistant strains of
Candida and Aspergillus spp. show reduced susceptibility to
voriconazole. | [General Description]
Voriconazole is a synthetically prepared, broad-spectrum triazole derivative of fluconazole, which shows in vitro activity against many yeasts and a broad-spectrum of mold and dermatophyte isolates. Its mode of action involves the inhibition of cytochrome P450 (CYP)-dependent enzyme, 14-α-sterol demethylase, and hence it is involved in disrupting the cell membrane and terminate the fungal growth. | [Pharmaceutical Applications]
A synthetic triazole formulated for oral and parenteral use. | [Biological Activity]
Triazole antifungal agent. Displays potent activity against Candida , Cryptococcus and Aspergillus species. | [Biochem/physiol Actions]
Voriconazole is an antifungal used to treat serious fungal infections. Voriconazole inhibits ergosterol synthesis by inhibiting CYP450-dependent 14-α sterol demethylase resulting in a depletion of ergosterol in fungal cell membranes. | [Pharmacokinetics]
Oral absorption: 96%
Cmax 400 mg oral: c. 2 mg/L after 2 h
Plasma half-life: c. 6 h
Volume of distribution: 4.6 L/kg
Plasma protein binding: 58%
Absorption
Oral absorption is rapid and almost complete, and is unaffected by intragastric pH. In adults, there is a disproportionate increase in blood concentrations with increasing oral and parenteral dosage, due to partial saturation of first-pass metabolism. In children given low dosages of the drug, proportional changes in drug levels are seen.
Distribution
It is widely distributed into body tissues and fluids, including brain and CSF.
Metabolism and excretion
It is extensively metabolized by the liver. More than 80% of a dose appears in the urine, but less than 2% is excreted in unchanged form. It is metabolized by several different hepatic cytochrome P450 enzymes. Some people with point mutations in the genes encoding these enzymes are poor metabolizers while others are extensive metabolizers. Drug levels are as much as four-fold lower in individuals who metabolize the drug more extensively. | [Clinical Use]
Acute and chronic invasive aspergillosis
Serious invasive Candida infections
Serious infections caused by Scedosporium and Fusarium spp. | [Side effects]
Unwanted effects include mild to moderate visual disturbance,
rashes, and transient abnormalities of liver enzymes.
Rare side effects include life-threatening hepatitis. | [Synthesis]
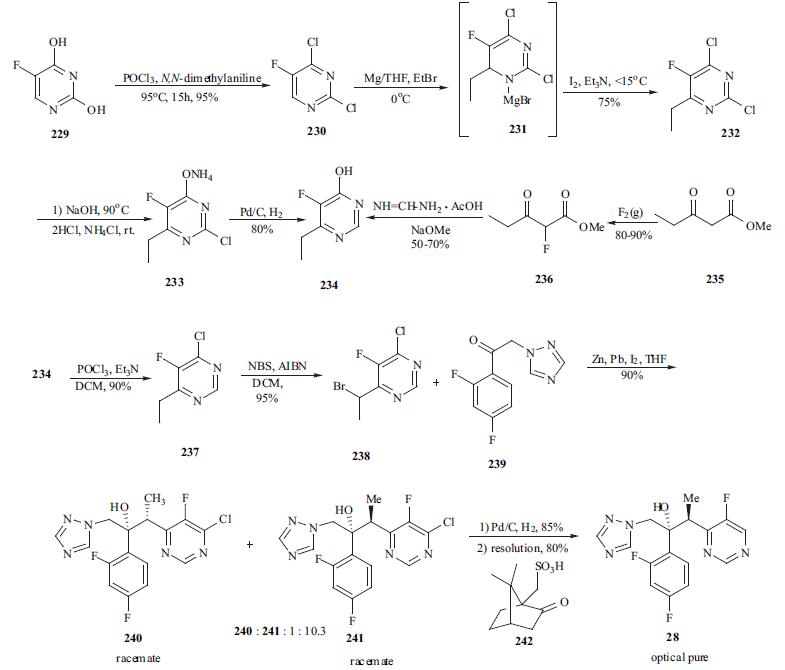
The synthesis of
voriconazole is an excellent example of process research. As
depicted in the scheme, 5-fluorouracil (229) was chlorinated
in both the 2- and 4- positions using a mixture of
phosphorus oxychloride and N,N-dimethylaniline at 95° C to
afford 230 in 95% yield. Dichloro pyrimidine 230 was
reacted with ethyl magnesium bromide to give
dihydropyrimidine adduct 231. Adduct 231 was oxidized
prior to quenching using a mixture of iodine and TEA in
THF to give 2,4-dichloro-6-ethyl-5-fluoro pyrimidine (232)
in 75% yield. Reaction of 232 with two equiv of aqueous
NaOH at reflux gave selective displacement of the chloro
functionality at 4-position. Acidification of the reaction and
extraction with DCM gave 2-chloro-6-ethyl-5-fluoro-4(3H)-
pyrimidine which was conveniently isolated as its ammonia
salt 233. Dechlorination of 233 was achieved using catalytic
hydrogenation at 50℃ to provide 234 in 80% yield.
Alternatively, 4-fluoro-6-ethyl-5-fluoropyrimidine (234) was
prepared in a two-pot process in which methyl 3-
oxopentanoate (235) was fluorinated with fluorine gas to
give methyl 2-fluoro-3-oxopentanoate (236) in 80-90% yield. This ester was then cyclized with formamidine
acetate in the presence of NaOMe to give 234 in a moderate
yield (50-70%). Reaction of 234 with phosphorus
oxychloride and TEA afforded 4-chloro-6-methyl-5-
fluoropyrimidine (237) in 90% yield. Reaction of 237 with
NBS in the presence of AIBN initiator provided bromide
238 in 95% yield. A Reformatsky protocol was employed in
the condensation of 238 with ketone 239 which was an
intermediate in the commercial synthesis of Diflucan. A
solution of iodine in THF was added to a slurry of zinc and
lead at rt and then a mixture of bromide 238 and ketone 239
were added to the above mixture at 5°C for 30 min. This
provided the best diastereomeric selectivity and the ratio of
241 and 240 enantiomeric pair reached approximately 10 to
1. Adduct 241 was de-chlorinated using standard
hydrogenation condition (5% w/w Pd on carbon /15 psi hydrogen) to give the racemate of voriconazole. The racemic
voriconazole was resolved using (1R)-10-camphorsulfonic
acid (242) and crystallization of the required diastereomeric
salt provided optically pure voriconazole (28) in 80% yield.
 | [Veterinary Drugs and Treatments]
Voriconazole may be a useful treatment for a variety of fungal infections
in veterinary patients, particularly against Blastomyces,
Cryptococcus, and Aspergillus. It has high oral bioavailability in a
variety of species and can cross into the CNS. Currently available
human dosage forms are extremely expensive, however, and little
clinical experience has occurred using voriconazole in veterinary
patients. There is considerable interest in using voriconazole for
treating aspergillosis in pet birds as their relative small size may
allow the drug to be affordable; additional research must be performed
before dosing regimens are available. | [Drug interactions]
Potentially hazardous interactions with other drugs
Analgesics: concentration of diclofenac, ibuprofen,
alfentanil, methadone and oxycodone increased,
consider reducing alfentanil and methadone dose;
concentration of fentanyl possibly increased.
Anti-arrhythmics: avoid with dronedarone.
Antibacterials: concentration reduced by rifabutin;
increase dose of voriconazole from 200 to 350 mg
and from 100 to 200 mg (depends on patient’s
weight), and increase IV dose to 5 mg/kg if used in
combination - avoid concomitant use if possible;
increased rifabutin levels - monitor for toxicity;
concentration reduced by rifampicin - avoid.
Anticoagulants: avoid with apixiban and rivaroxaban;
enhanced effect of coumarins.
Antidepressants: avoid concomitant use with
reboxetine; concentration reduced by St John’s wort
- avoid.
Antidiabetics: possibly increased concentration of
sulphonylureas.
Antiepileptics: concentration possibly reduced
by carbamazepine, phenobarbital and primidone
- avoid; fosphenytoin and phenytoin reduces
voriconazole concentration and voriconazole
increases fosphenytoin and phenytoin concentration
- double oral voriconazole dose and increase IV to 5
mg/kg dose if using with phenytoin; avoid if possible.
Antimalarials: avoid concomitant use with
artemether/lumefantrine and piperaquine with
artenimol.
Antipsychotics: concentration of lurasidone
increased - avoid concomitant use; increased risk
of ventricular arrhythmias with pimozide - avoid
concomitant use; possibly increased quetiapine levels
- avoid concomitant use.
Antivirals: concentration increased or decreased by
atazanavir and concentration of atazanavir reduced;
concentration of daclatasvir possibly increased -
reduce daclatasvir dose; concentration possibly
affected by darunavir; concentration reduced by
efavirenz and ritonavir; also concentration of
efavirenz increased - avoid with ritonavir; with
efavirenz reduce dose by 50
% and increase dose of
voriconazole to 400 mg twice daily; concentration
possibly increased by simeprevir - avoid;
concentration possibly affected by telaprevir -
increased risk of ventricular arrhythmias; possibly
increased saquinavir levels; concentration of
simeprevir possibly increased - avoid.
Avanafil: possibly increased avanafil concentration -
avoid.
Benzodiazepines: may inhibit metabolism of
diazepam and midazolam.
Ciclosporin: AUC increased - reduce ciclosporin
dose by 50
% and monitor closely.
Clopidogrel: possibly reduced antiplatelet effect.
Cytotoxics: possibly increases bosutinib
concentration - avoid or reduce dose of bosutinib;
possibly increases crizotinib and everolimus
concentration - avoid; possibly increases ibrutinib,
pazopanib and ponatinib concentration - reduce
dose of ibrutinib, pazopanib and ponatinib; avoid
with ceritinib, lapatinib, nilotinib, cabazitaxel and
docetaxel (or reduce dose of cabazitaxel, ceritinib
and docetaxel); reduce dose of panobinostat and
ruxolitinib.
Domperidone: possible increased risk of arrhythmias
- avoid.
Ergot alkaloids: risk of ergotism - avoid.
Ivacaftor and lumacaftor: possibly increase ivacaftor
concentration - reduce dose of ivacaftor and ivacaftor
with lumacaftor.
Lipid-lowering drugs: possibly increased risk of
myopathy with atorvastatin or simvastatin; avoid
with lomitapide.
Ranolazine: possibly increased ranolazine
concentration - avoid.
Retinoids: possibly increased risk of tretinoin
toxicity.
Sirolimus: increased sirolimus concentration - avoid.
Tacrolimus: AUC increased - reduce tacrolimus
dose to a third and monitor closely.
Ulcer-healing drugs: esomeprazole and omeprazole
concentration increased - reduce omeprazole dose
by 50
%. | [Metabolism]
Voriconazole is metabolised by hepatic cytochrome P450
isoenzyme CYP2C19; the major metabolite is the inactive
N-oxide. Metabolism via isoenzymes CYP2C9 and
CYP3A4 has also been shown in vitro.
Voriconazole is eliminated via hepatic metabolism with
less than 2
% of the dose excreted unchanged in the urine.
After administration of a radiolabelled dose of
voriconazole, approximately 80
% of the radioactivity
is recovered in the urine as metabolites. The majority
(>94
%) of the total radioactivity is excreted in the first 96
hours after both oral and intravenous dosing | [storage]
Store at -20°C | [Mode of action]
Voriconazole is structurally related to fluconazole (Pfizer, diflucan) and acts by inhibiting the cytochrome P450- dependant enzyme 14a-sterol demethylase of ergosterol synthesis (thereby resulting in the formation of a cell membrane with abnormal characteristics and accumulation of toxic sterol intermediates). Voriconazole was more active than itraconazole and fluconazole against Cryptococcus neoformans and a variety of Candidas species such as C. albicans, C. glabrata C. krusei. It also exhibits similar or superior activity compared to amphotericin B and itraconazole against filamentous fungi such as Aspergillus, an important pathogen which is not susceptible to fluconazole. | [Precautions]
Significant drug interactions include cyclosporins(increased cyclosporine levels), phenytoin, rifampin,and rifabutin (decreased voriconazole levels). Becauseof its low toxicity profile, this drug may gain importancein the chronic treatment of infections with invasive dimorphicfungi and resistant Candida spp. | [References]
[1] sabo ja, abdel-rahman sm. voriconazole: a new triazole antifungal. ann pharmacother. 2000 sep;34(9):1032-43.
[2] johnson lb, kauffman ca. voriconazole: a new triazole antifungal agent. clin infect dis. 2003 mar 1;36(5):630-7. |
|
|