背景及概述[1]
1-甲基-2-吡咯乙酸甲酯可用作医药合成中间体,可由N-甲基吡咯和乙二酰氯为反应原料,先制备中间体2-(1-甲基-1H-吡咯-2-基)-2-氧代乙酸,进一步反应制备2-(1-甲基-1H-吡咯-2-基)乙酸,最后在对甲苯磺酸的作用下酯化生成1-甲基-2-吡咯乙酸甲酯。
制备[1]
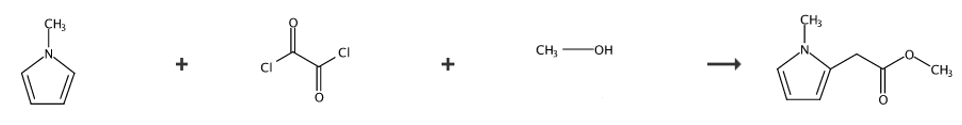
第1步:2-(1-甲基-1H-吡咯-2-基)-2-氧代乙酸
将乙二酰氯(4.30mL,50mmol)和CH2Cl2(20mL)加入三口圆-底部烧瓶,并在氮气氛下冷却至-10℃,经由滴液漏斗逐滴添加在CH2Cl2(40mL)中的N-甲基吡咯(4.44mL,50mmol),保持温度低于0℃,将所得溶液在0℃下搅拌1小时,然后添加足够的20%KOH(水溶液)以使溶液的pH为10,将溶液搅拌30分钟,然后用H2O稀释。分离两层,水层用CH2Cl2萃取,向合并的有机层中加入20%H2SO4(水溶液),直到形成白色沉淀,并将溶液搅拌30分钟。过滤固体,用冷H2O洗涤,然后减压干燥,得到标题化合物,为浅黄色固体,产率5.28g,69%。mp:140-141℃。1HNMR(300MHz;CDCl3):δH=8.05(1H,dd,J=4.4,1.6Hz,CCH),7.10(1H,app.t,J=1.8Hz,CHN),6.28(1H,dd,J=4.4,2.4Hz,CHCHN),3.99(3H,s,NCH3);13CNMR(75MHz,CDCl3):δC170.5、160.5、136.7、128.9、126.7、111.1、38.3;
第2步:2-(1-甲基-1H-吡咯-2-基)乙酸
2-(1-甲基-1H-吡咯-2-基)-2-氧乙酸(3.08g,20mmol),一水合肼(2.00mL,40mmol)和20%KOH(aq)(40mL)在氮气气氛下添加到装有回流冷凝器的两颈圆底烧瓶中,将得到的溶液在100℃下回流八小时,冷却至室温后,加入2MHCl以使溶液的pH为2,将混合物用CH2Cl2萃取,并将有机物用H2O洗涤,然后用MgSO4干燥并过滤。减压除去溶剂,并将粗品从Et2O和汽油中重结晶,得到标题化合物,为黄色固体,产率1.88g,68%。mp:107-108℃。1HNMR(300MHz;CDCl3):δH=6.61(1H,app.t,J=2.3Hz,CHN),6.10-6.06(2H,m,CHCHN,CCH),3.67(2H,s,CH2CO2H),3.59(3H,s,NCH3);13CNMR(75MHz,CDCl3):δC176.9,124.1,122.9,109.3,107.3,34.0,32.4;m/z。
第3步:1-甲基-2-吡咯乙酸甲酯
在氮气气氛下,将2-(1-甲基-1H-吡咯-2-基)乙酸(1.14g,8mmol)和对甲苯磺酸(0.47g,2mmol)加入装有回流冷凝器的两颈圆底烧瓶中,加入MeOH(20mL),反应在70℃下回流八小时。冷却至室温后,减压除去甲醇,并将粗产物用CH2Cl2稀释,然后用盐水洗涤。将有机物用MgSO4干燥,过滤,并在减压下浓缩。粗产物通过快速柱色谱纯化(汽油:EtOAc(90∶10),Rf=0.38),得到标题化合物1-甲基-2-吡咯乙酸甲酯,为无色油,具有根据文献的光谱数据,收率0.98g,80%。1HNMR(300MHz;CDCl3):δH=6.60(1H,app.t,J=2.1Hz,CHN),6.09-6.04(2H,m,CCH,CHCHN),3.71(3H,s,NCH3),3.64(2H,s,CH2CO2Me),3.58(3H,s,OCH3);13CNMR(75MHz,CDCl3):δC171.1,124.9,122.7,108.9,107.1,52.2,34.0,32.6。
参考文献
[1] James E. Taylor, Matthew D. Jones, Jonathan M. J. Williams,等. Friedel-Crafts acylation of pyrroles and indoles using 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) as a nucleophilic catalyst[J]. Organic Letters, 2015, 42(13):5740-3.