头孢氨苄(Cephalexin,CEX)又称头孢立新、头孢菌素Ⅳ,其化学名为(6R,7R)-3-甲基-7-[(R)-2-氨基-2-苯乙酰氨基]-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸,化学式C16H17N3O4S,属于代口服类头孢菌素类抗生素药物。于1967年由美国礼莱公司开发,1970年投放市场。由于其抗菌谱广、生物利用率高、耐受性强,头孢氨苄已经成为目前应用最广的头孢类抗生素品种。
头孢氨苄主要通过抑制细胞壁的合成,从而使细胞内容物过度生长膨胀至破裂,导致细胞内容物外泄,杀死细菌。其具有抗菌谱广、杀菌力强、耐酸、胃肠吸收好等优点,口服吸收后分布良好,一小时可达血药浓度峰值,但吸收量不减,能投入各种组织中。除了肠球菌素和耐甲氧西林葡萄球菌外,肺炎链球菌、溶血性链球菌、产或不产青霉素酶葡萄球菌等大部分临床常见革兰阳性致病菌对该药敏感。该药主要用于治疗呼吸道感染、尿路感染和皮肤软组织感染等。
【理化性质】
头孢氨苄常温下为白色或微黄色结晶性粉末,微臭,在水中微溶,在氯仿、乙醇或乙醚中不溶,熔点为326.81℃,沸点为636.05℃,pKa为2.5、5.2、7.3,其水溶液(5mg/mL)比旋度为+149°~+158,pH为3.5~5.5,在262nm紫外吸收区有吸收波长。头孢氨苄在干燥状态下较稳定,但对紫外光较敏感,并由于含有内酰胺环,在碱性(pH>9)或β-内酰胺酶存在条件下易水解开环。在干燥状态及固态下相对比较稳定,但遇强酸、强碱、高热和紫外线均易分解;在酸碱环境中的稳定性:在以下的水溶液中较稳定,但在以上的水溶液中则迅速被破坏。
【作用机理】
头孢氨苄为广谱抗生素类药物,在人体内的作用机理是通过抑制细胞壁的合成,使细胞里的物质膨胀最后破裂溶解,从而达到杀菌作用。人类的细胞无细胞壁,因此对人是无害的。主要用于革兰氏阳性菌和阴性菌感染,对于耐青霉素的菌类也有很好的杀菌效果。其进入体内后吸收迅速而完全,生物利用率高。
【药理作用】
头孢氨苄的抑菌机理是通过抑制细菌细胞壁的合成,使细胞内容物膨胀至细胞自溶,从而达到杀菌作用。头孢氨苄是广谱抗生素。对革兰氏阳性菌及革兰氏阴性菌均具有抗菌作用,其抗菌能力比青霉素类大20倍,比磺胺类大10倍、比喹诺酮类大5倍。 头孢氨苄为代头孢菌素,其抗菌谱与头孢噻吩基本相同,但抗菌活性比后者弱。除肠球菌属、耐甲氧西林金黄色葡萄球菌外,肺炎链球菌、溶血性链球菌、产或不产青霉素酶葡萄球菌的大部分菌株对该品敏感。头孢氨苄对奈瑟菌属抗菌作用明显,但对流感嗜血杆菌的作用不显著;该品对部分大肠埃希菌、奇异变形杆菌、沙门菌和志贺菌具有一定的抗菌作用。其余肠杆菌科细菌、不动杆菌、铜绿假单胞菌、脆弱拟杆菌均对该品呈现耐药性。梭杆菌属和韦容球菌一般对该品敏感,厌氧革兰氏阳性球菌对该品中度敏感。
【药代动力学】
头孢氨苄口服吸收良好,血药峰浓度(Cmax)在患者空腹口服该品0.5g,1小时后可以达到,Cmax平均为18mg/L。餐后服药不仅不会减少其吸收量还可以延长吸收时间并降低Cmax。患有幼儿乳糜泻和小肠憩室的患者会增加对该品的吸收,而患有克隆病和肺囊性纤维化的患者会延缓和减少对该药品的吸收。老年人的胃肠道的吸收量较年轻人来说并没有减少,却会维持更久的Cmax。其血消除半衰期(t1/2b)为0.6~1.0h,如加服丙磺舒可以提高其血药浓度,并且血消除半衰期可延长至1.8h;肾衰竭患者的t1/2b可延长至5~30小时;新生儿的血消除半衰期可达6.3h。
头孢氨苄进入体内后广泛分布于各组织体液中,每6小时口服0.5g时,痰液中平均浓度为0.32mg/L,其在脓痰中的浓度较高。脓液药物浓度基本与血药浓度相等,关节腔渗出液中的药物浓度为血药浓度的50%。 头孢氨苄会透过胎盘进入胎儿血循环及产妇羊水;哺乳期妇女口服0.5g后,乳汁浓度为5mg/L。约有5%的口服给药量由胆汁排出,故胆汁中药物浓度为血药浓度的1~4倍。血清蛋白结合率为10%~15%。该品不在体内代谢,通过尿液排出,24小时内排出给药量的80%~90%,口服0.5g后尿药峰浓度可达2.2g/L。血液透析和腹膜透析可以清除头孢氨苄。 头孢氨苄是半合成的代口服给药的头孢霉素,抗菌谱与头孢噻吩、头孢噻啶基本相同,但抗菌作用较后两者弱,但其的特点是耐酸,并且口服给药吸收良好。对耐药金黄色葡萄球菌有良好的抑制作用。
【制备方法】
1.化学半合成法
(1)7-ADCA四甲基胍盐的合成 连接反应装置,在干燥洁净的反应瓶中加入二氯甲烷50mL,7-ADCA20g,开启搅拌,开启冷泵降温,冷却至-10~-5℃。向反应瓶中加入四甲基胍11.2g,控制反应温度不高于10℃,搅拌、溶解后,将料液温度降至-25℃。得到7-ADCA四甲基胍盐的二氯甲烷溶液,待用。
(2)混合酸酐的制备 连接反应装置,向干燥洁净的反应瓶中加入二氯甲烷100mL,左旋苯甘氨酸邓钾盐31.5g,N-甲基乙酰胺0.8g,将混合料液温度降温至-35℃,加入特戊酰氯13g,4-甲基吡啶两滴。然后升温至-25℃,搅拌反应2h,降温至-65℃。
(3)缩合反应 将制备好的7-ADCA的四甲基胍盐的二氯甲烷溶液转移至制备好的2.2.2反应瓶中,在-30℃左右反应3h(反应2.5h取样液相检测,以反应液中7-ADCA残留量不超过1%为反应终点,7-ADCA残留量大于1%时可适当延长反应时间,但是最长缩合反应时间不应超过3.5h)。
(4)水解 向水解反应瓶中加入去离子水水120g,调节去离子水的温度至10℃,加入30%盐酸28g,将缩合液倒入水解瓶中,充分搅拌20min。静置20min,分去二氯甲烷层,有机相回收。水层加入2g三乙胺,调节pH值至2.0~2.2,过滤,少量水洗涤后合并滤液,将料液转移至结晶瓶中。
(5)析晶干燥 向结晶瓶中加入三乙胺调节pH值至2.8左右,养晶30min,晶体析出后继续用三乙胺调节pH值至5.0.冷却至0~5℃,调节搅拌速度至合适速度搅拌3h过滤,用60mL50%丙酮/水的混合液洗涤滤饼,抽干后用40mL纯丙酮洗滤饼,抽干。将固体产品转移至真空干燥箱中,在(45±5)℃真空干燥4h,得到头孢氨苄-水合物33g,含量≥96.4%,水分3.6%,7-ADCA残留≤0.06%,总收率(摩尔)≥91%。
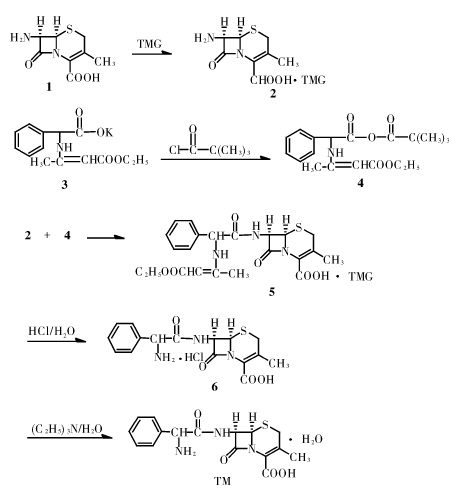
图1为头孢氨苄的合成路线图
2.酶半合成法
(1)酶催化合成 在合成反应罐中加入一定的合成酶;在溶解罐中溶解7-ADCA,pH=7.5;将7-ADCA转移至合成罐,PGM溶解液采用滴加方式,用浓NH3?H2O控制pH。加完后用6mol/L盐酸和3mol/L氨水控制反应pH,用循环冷冻机控制反应温度,通过检测7-ADCA的转化率判断反应终点,转化率大于98%以上终止反应。
(2)分离 通过反应器底部筛网进行头孢氨苄和固体酶的分离,用母液循环洗涤直至母液无白色物质。
(3)重结晶 滤饼溶于母液中,用浓盐酸溶解,温度20℃;溶解液过100μm滤纸后转移至结晶罐,用3mol/L氨水调晶。一调PH=2.2~2.5,T=30~35℃,养晶30min;二调至PH=4.8~5.0,养晶3h。
(4)抽滤、丙酮洗涤、45℃干燥2h后得头孢氨苄成品。
【应用】
头孢氨苄主要用于敏感菌所导致的呼吸道感染、泌尿道感染、妇产科感染、皮肤及软组织感染、淋病等。
【主要参考资料】
[1]张伟. 头孢氨苄单克隆抗体的研制及免疫学检测方法的初步建立[D].扬州大学,2017.
[2]李淑桃. pH响应再生型两水相体系相转移催化合成头孢氨苄[D].华东理工大学,2014.
[3]陆莹莹. 酶法合成头孢氨苄的反应—膜萃取耦合过程[D].天津大学,2014.
[4]赵启法. 头孢氨苄人工抗原的合成及单克隆抗体的制备[D].郑州大学,2012.
[5]王艳艳,袁国强,朱科,王进贤.酶法合成头孢氨苄工艺研究[J].中国抗生素杂志,2013,38(07):516-519.
[6]李文杰,赵南,胡文彩.头孢氨苄工艺改进[J].广州化工,2016,44(02):74-75+78.
[7]安蔚. 酶法合成头孢氨苄过程中固定化青霉素酰化酶酶活稳定性及其再生研究[D].河北科技大学,2016.