背景及概述
3-氮杂双环[3.1.0]己烷类衍生物是一种氮杂环化合物,它是一类具有独特生物活性的一类分子,常用作医药和农药的结构单元。含氮化合物较易于结构的修饰,因此可以很方便地引入各种基团,使其具有不同的性能。(1alpha,5alpha,6alpha)-3-氮杂双环[3.1.0]己烷-3,6-二甲酸3-叔丁酯英文名称:(1R,5S,6r)-3-(tert-Butoxycarbonyl)-3-azabicyclo[3.1.0]hexane-6-carboxylicacid,CAS号:927679-54-7,分子式:C11H17NO4,分子量:227.257,密度:1.276,沸点:355.9ºCat760mmHg。
制备
目前国内外对3-氮杂双环[3.1.0]己烷的合成已经有不少报道,3-氮杂双环[3.1.0]己烷主要有两部分构成,一个五元氮杂环和一个三元环。因此在制备上也离不开五元环和三元环的构建。五元环常在顺式-1,2-二取代环丙烷的基础上构建;三元环通常由共轭五元环与卡宾或硫叶立德的反应。(1ALPHA,5ALPHA,6ALPHA)-3-氮杂双环[3.1.0]己烷-3,6-二甲酸3-叔丁酯的合成以较为廉价的富马酸二乙酯和氯乙酸乙酯为起始原料,通过碱催化下的迈克尔加成反应构建环丙烷结构,文献报道的产率达到了80%,后续反应条件都比较温和,且产率均比较高。在制备I-3的时候,按照文献报道,在酯基和酰胺键同时存在的情况下可以选择性地将酰胺键还原,酯基保留,而且产率高达90%,整条路线仅步反应产率80%,其他各步均在85%以上,符合大规模工业生产的要求[1]。制备路线图如下:
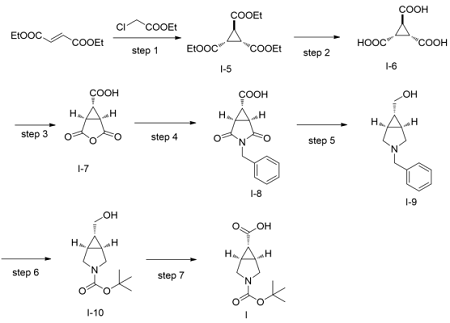
图1(1ALPHA,5ALPHA,6ALPHA)-3-氮杂双环[3.1.0]己烷-3,6-二甲酸3-叔丁酯合成路线图
1,2,3-三甲酸乙酯环丙烷的合成(I-5)
在装有温度计,回流冷凝管的5L四口瓶中,机械搅拌下加入2.5LN,N-二甲基甲酰胺,1603g碳酸钾(11.62mol,4.0eq),6.6g苄基三乙基氯化铵(29mmol,0.01eq),加热至40℃,缓慢滴加含有500g富马酸二乙酯(2.9mol,1.0eq)和477.7g氯乙酸乙酯(3.92mol,1.35eq),滴毕,保持40℃反应,48h后反应完全。反应液静置2h,上清液浓缩,尽可能将DMF旋去。向滤饼中加入1L乙酸乙酯,抽滤,滤饼用200ml乙酸乙酯洗涤,合并有机相以及之前的浓缩液,加入1L水,静置分层,取上层,水层继续用750mL乙酸乙酯萃取(250mL*3次),合并有机相,无水硫酸钠干燥,过滤,滤液浓缩至干,得到黑褐色油状物,减压蒸馏,收集125~130℃馏分557g,收率75.5%。
1,2,3-三甲酸环丙烷的合成(I-6)
在装有温度计,回流冷凝管的2L四口瓶中,加入295g氢氧化钠(7.38mol,3.3eq),750mL水,机械搅拌下待溶液降至室温,加入250mL乙醇,将反应液升温至50℃,滴加529.9g1,2,3-三甲酸乙酯环丙烷(2.05mol,1.0eq),控制滴加速度,使反应液低于回流温度,滴毕,升温回流2h,反应完全。将反应液冷至室温,将反应液中乙醇旋掉。所得的反应液在冰浴下,缓慢滴加670mL浓盐酸(8.12mol,3.63eq),此时反应液pH=1,将反应液旋干,得到白色固体,加入1L丙酮回流,稍冷,过滤,滤饼用200mL丙酮洗一遍,滤液旋干,得到347g白色固体,产率98.5%,
(1R,5S,6S)-2,4-二氧代-6-羧酸-3-氧杂二环[3.1.0]己烷的合成(I-7)
在装有温度计,回流冷凝管的2L三口瓶中,加入347g底物(1.99mol,1.eq)机械搅拌下加入,1L冰醋酸,268g醋酸酐(2.63mol,1.32eq),升温回流3h,反应完全。将反应液中的冰醋酸以及过量的乙酸酐旋干,所得灰白色固体,加入300mL甲苯打浆,过滤,真空烘箱中干燥,得到白色固体302.1g,收率97.1%,
(1R,5S,6R)-3-苄基-6-甲酸-2,4-二氧代-3-氮杂双环[3.1.0]己烷(I-8)
在装有温度计,分水器,回流冷凝管的2L四口瓶中,加入300g底物(1.92mol,1.0eq),机械搅拌下加入500mL丙酮,冰浴下缓慢滴加216g苄胺(2.02mol,1.05mol),滴毕,升温蒸去丙酮,继续升温至160~180℃,反应过程中观察到水珠的生成,直到观察不到水珠为止,停止反应。待反应液冷却至60℃,向三口瓶中倒入1L水,10mL浓盐酸,析出大量白色固体,抽滤,滤饼烘干,得到436g白色固体,收率92.6%,
(1R,5S,6R)-3-苄基-6-甲酸乙酯-2,4-二氧代-3-氮杂双环[3.1.0]己烷的合成(I-2)
在装有温度计,回流冷凝管的250ml四口瓶中,加入10.0g底物(41mmol,1.0eq),机械搅拌下加入50mL乙醇,加热至60℃使固体全部溶解,缓慢滴加10g浓硫酸(102mmol,2.5eq),反应6h完全。将反应液冷却至室温,有白色固体析出,抽滤,滤饼用少量冷乙醇洗涤,烘干,得白色鳞片状固体8.36g,收率75.2%。
(1R,5S,6R)-3-苄基-6-甲醇-3-氮杂双环[3.1.0]己烷的合成(I-9)
在装有温度计,回流冷凝管的2L三口瓶中,机械搅拌下加入100g底物(0.41mol,1.0eq),1L四氢呋喃,冰浴下分批加入78g硼氢化钠(2.05mol,5.0eq),继续保持冰浴下缓慢滴加291.3g三氟化硼乙醚(2.05mol,5.0eq),滴完后继续保温1h,撤去冰浴,室温下保持1h,缓慢升温回流6h,反应完毕。将反应液在冰浴下冷却,将反应液向600mL冰水中倾倒,产生大量气泡,淬灭完后加入150mL浓盐酸(1.8mol,4.5eq),转移到2L三口瓶中回流5h,将反应液稍冷,将瓶中四氢呋喃尽可能旋去,此时瓶中有固体析出,过滤,滤液冷却至室温,用50%氢氧化钠调pH=9,600mL乙酸乙酯萃取3次(200mL*3),合并有机相,无水硫酸钠干燥,浓缩,得到淡黄色粘稠液体,减压蒸馏,收集146~150℃馏分72.9g,收率88.1%。在装有温度计,回流冷凝管的1L三口瓶中,机械搅拌下加入600mL四氢呋喃,冰浴下,分批加入23.3g四氢铝锂(612mmol,5.0eq),加完后继续保持冰浴,分批加入30g底物(122mmol,1.0eq),加完后,继续搅拌半小时,撤去冰浴,搅拌1h,缓慢升温回流6h,反应完全。将反应液在冰浴下冷却,一次分别缓慢滴加25mL水,25mL15%氢氧化钠溶液,75mL水,抽滤,滤饼用150mL四氢呋喃洗涤一遍,浓缩,得到31.6g淡黄色粘稠液,减压蒸馏,收集146~150℃馏分22.8g,收率92.1%。
(1R,5S,6R)-3-叔丁氧羰基-6-甲醇-3-氮杂双环[3.1.0]己烷的合成(I-10)
向含有磁转子的500mL高压反应釜中加入26.5g底物(138.5mmol,1.0eq),250mL甲醇,5.5g10%钯炭,60.4g二碳酸二叔丁酯,用N2将反应釜中的空气排空,重复3次,再用水泵抽真空。往高压反应釜中通H2至2.0MPa,升温至45℃反应,当H2吸完后,继续补加H2,直到高压反应釜中气压恒定。反应完后将反应液过滤,用50mL甲醇洗涤滤饼,滤液浓缩,得到23.6g淡黄色粘稠液体,收率85.3%,不经过纯化直接进行下一步反应。
(1R,5S,6R)-3-叔丁氧羰基-6-甲酸-3-氮杂双环[3.1.0]己烷的合成(I)
在装有温度计,回流冷凝管的250mL四口瓶中,机械搅拌下加入5.0g底物(23.4mmol,1.0eq),50mL乙腈,50mLpH=6.7的磷酸二氢钠-磷酸氢二钠缓冲溶液,0.18gTEMPO(1.17mmol,0.05eq),升温至35℃,同时滴加含有5.3g80%的亚氯酸钠(46.9mmol,2.0eq)的30mL溶液和含有1mL5%次氯酸钠(1.17mmol,0.05eq)的10mL溶液,控制滴加速度在2h左右滴完,继续反应4h结束。将反应液在冰浴下冷却,用20%氢氧化钠调节pH=8,加入含12g亚硫酸氢钠的50mL溶液(该步骤放热,需要在冰浴下进行),搅拌0.5h,用50mL二氯甲烷洗涤水层,弃去有机层,水层用2N稀盐酸调pH=3,加入60mL乙酸乙酯,静置分层,取有机相,水相继续用50mL乙酸乙酯萃取一次,合并有机相,无水硫酸钠干燥,浓缩,得到4.6g白色固体,收率88.3%。
参考文献
[1] Kozhushkov L D.[J]. Synthesis,2003, 6: 956-958.