R-(-)-度洛西汀
| 中文名称 | R-(-)-度洛西汀 |
|---|---|
| 中文同义词 | (R)-(-)-N-甲基-3-(1-萘氧基)-3-(2-噻吩)-丙胺;R-(-)-度洛西汀;右旋度洛西汀盐酸盐;R-度洛西汀盐酸盐;度洛西汀杂质A(单体);R-度洛西汀草酸盐;(-)-度洛西汀;盐酸度洛西汀杂质VII |
| 英文名称 | (R)-Duloxetine |
| 英文同义词 | (r)-duloxetine;(r)-n-methyl-gama-(1-naphthalenyloxy)-2-thiophenepropanamine;R-DULOXETINE HCL;(R)-N-Methyl-3-(naphthalen-1-yloxy)-3-(thiophen-2-yl)propan-1-aMine hydrochloride;N-methyl-3-(1-naphthalenyloxy)-3-thiophen-2-yl-1-propanamine;Ariclaim;CYMBALTA; ARICLAIM;2-Thiophenepropanamine, N-methyl-γ-(1-naphthalenyloxy)-, (γR) |
| CAS号 | 116539-60-7 |
| 分子式 | C18H19NOS |
| 分子量 | 297.41 |
| EINECS号 | 1312995-182-4 |
| 相关类别 | 中间体;医药原料;原料;原料药;原料;Pharmaceutical intermediate |
| Mol文件 | 116539-60-7.mol |
| 结构式 | 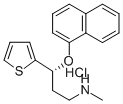 |
R-(-)-度洛西汀 性质
| 沸点 | 466.2±40.0 °C(Predicted) |
|---|---|
| 密度 | 1.158 |
| 酸度系数(pKa) | 10.02±0.10(Predicted) |
度洛西汀(duloxetine)是2002年上市的第三代抗抑郁药,临床使用其盐酸盐,商品名Cymbalta(欣百达)。由美国礼来公司(Elililly)研发上市。度洛西汀是一种新型的5-羟色胺(5-HT)和去甲肾上腺素(NE)高度特异性的双重抑制剂,通过抑制神经元突触对5-羟色胺(5-HT)和去甲肾上腺素(NE)的再摄取,增加神经递质水平,从而显著改善抑郁患者的情绪不适症状和疼痛性躯体症状而产生抗抑郁作用。
本信息由Chemicalbook晓楠编辑。盐酸度洛西汀(duloxetine hydrochloride)是新的选择性5-羟色胺和去甲肾上腺素再摄取双重作用的抑制剂,具有抗抑郁作用,同时对中枢性疼痛有抑制作用。药理学特点是能抑制神经元突触前膜对5-羟色胺和去甲肾上腺素的再摄取,对多巴胺再摄取的抑制作用较低。盐酸度洛西汀的适应症是抑郁症,它对内源性和非内源性抑郁、以及抑郁症伴发的疼痛症状都有效。治疗剂量为60mg/d~120mg/d,安全性好,不良反应少,较常见的不良反应有恶心、口干、便秘、食欲低下、疲劳、嗜睡和出汗增多。另一个适应证是糖尿病性神经病变所致的疼痛。
【商品名】 Cymbalta
【别 名】 LY-264453,LY-248686
【药效类别】 抗抑郁药,治疗紧张性尿失禁
【CAS】 136434-34-9
【研制单位】 日本Shionogi公司,美国Eli-Lilly公司
度洛西汀是一种选择性的5-HT与NE再摄取抑制剂(SSNRI)。度洛西汀抗抑郁与中枢镇痛作用的确切机制尚未明确,但认为与其增强中枢神经系统5-羟色胺与去甲肾上腺素能功能有关。临床前研究结果显示,度洛西汀是神经元5-HT与NE再摄取的强抑制剂。对多巴胺再摄取的抑制作用相对较弱。体外研究结果显示,度洛西汀与多巴胺能受体、肾上腺素能受体、胆碱能受体、组胺能受体、阿片受体、谷氨酸受体、GABA 受体无明显亲和力。度洛西汀不抑制单胺氧化酶。
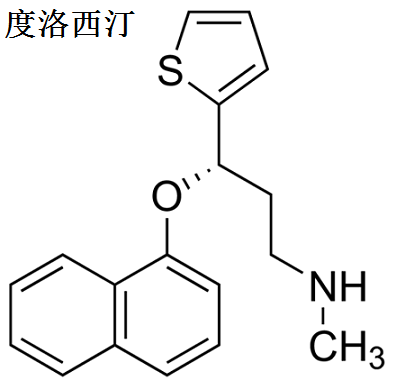
图1为度洛西汀分子结构式。度洛西汀口服后,半衰期大约为12小时(变化范围为8~17小时),在治疗范围之内其药代动力学参数与剂量成正比,一般服药3天后达到稳态血药浓度。度洛西汀主要经肝脏代谢,涉及两种P450酶:CYP2D6和CYP1A2。
口服度洛西汀后,平均滞后2小时,药物开始吸收(Tlag);口服6小时后度洛西汀达到最大血浆浓度(Cmax),进食不影响Cmax,但是将延迟达峰时间6~10小时,略微降低吸收程度,约10%。晚间一次服药与晨间一次服药相比,度洛西汀的吸收滞后3小时,表现清除增加1/3。
表观分布容积平均为1640升。度洛西汀与人体血浆蛋白有高度亲和性(大于90%),主要与白蛋白和α1-酸性糖蛋白结合。 目前为评价度洛西汀和其他高蛋白结合药物之间是否有药物相 互作用。肝或肾功能不全不影响度洛西汀的血浆蛋白结合。
度洛西汀在人体内代谢广泛,代谢产物多。血浆中的代谢产物包括葡萄糖醛酸结合的4-羟基度洛西汀、硫酸结合的5-羟基-6-甲氧基度洛西汀。尿中分离出多种其他代谢产物,有些仅出现在小的消除代谢旁路中。尿液中仅有少量未经代谢的盐酸度洛西汀原型(1%),大部分(70%)以盐酸度洛西汀代谢产物形式经尿液排出,大约20%经粪便排出。用于治疗抑郁症。吞服,不要咀嚼和压碎。推荐起始剂量为40mg/d(40mg一日一次或20mg,一日两次)至60mg/d(一日一次),不考虑进食影响。1.禁用于已知对度洛西汀或产品中任何非活性成分过敏的患者。
2.禁止与单胺氧化酶抑制剂(MAOIs)联用,也不可以在MAOIs停药14天内使用本品;根据度洛西汀的半衰期,停用度洛西汀后至少5天,才能开始使用MAOIs。
3.临床显示度洛西汀有增加瞳孔散大的风险,因此未经控制的闭角型青光眼患者应避免使用度洛西汀。1.肝肾功能不全,会明显减低度洛西汀的代谢和清除,不建议用于该类患者。
2.度洛西汀与酒精交互作用可能会造成肝损伤,通常不建议用于治疗酗酒患者。
3.临床试验中,度洛西汀与安慰剂相比,收缩压平均升高2mmHg,舒张压平均升高0.5mmHg。建议在治疗前及治疗中定期测量血压。度洛西汀通过两种CYP2D6和CYP1A2代谢,中度抑制CYP2D6,但不抑制也不诱导CYP1A2和CYP3A4。与其他主要通过CYP2D6,且治疗窗狭窄的药物(如:TCAs、Ic类抗心律失常药物、吩噻嗪)时,应谨慎。【孕妇即哺乳期妇女用药】 妊娠分类C,怀孕妇女服用本品是否安全尚不明确。对于孕妇,应权衡利弊决定是否服用本品。只有当潜在利益高于风险才可以使用,否则怀孕期及哺乳期内不应使用。
【儿童用药】 目前尚缺乏在儿童中的足够临床经验。
【老年人用药】 临床研究中未观察到老年人群与年轻人群在安全性和疗效方面的显著差异,但不能排除某些老年患者的敏感性增高。度洛西汀的合成路线按照醚化反应所用原料的不同,主要可分为以下两大类:①以(S)-3-N,N-二甲氨基-1-(2-噻吩基)-1-丙醇67为原料;②以(S)-3-N-甲氨基-1-(2-噻吩基)-1-丙醇72为原料。
1. 以2-乙酰噻吩71为原料,在乙醇中与二甲胺盐酸盐69和多聚甲醛 70发生Mannich反应生成3-N,N-二甲氨基-1-(2-噻吩基)-1-丙酮68。酮叔胺68经硼氢化钠还原为醇叔胺67的消旋体,接着用(S)-(+)-扁桃酸进行拆分得到(S)-3-N,N- 二甲氨基-1-(2-噻吩基)-1-丙醇67。手性醇叔胺67在氢化钠作用下于二甲亚砜(DMSO)中与1-氟萘进行SNAr氧醚化反应,生成N-甲基度洛西汀65,再与氯甲酸苯酯(或氯甲酸三氯乙酯)反应生成度洛西汀的氨基甲酸酯88,最后不经分离直接水解就制得度洛西汀64。这是早期合成度洛西汀的一条最基本、技术相对成熟的合成路线,也比较适合于工业化生产。
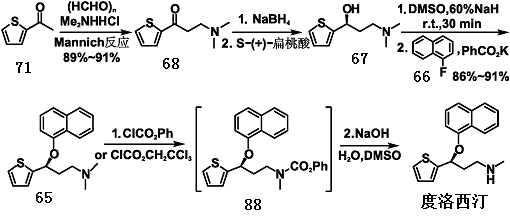
图2为以2-乙酰噻吩为原料合成度洛西汀的路线图。
2. 以2-乙酰噻吩71为起始原料,在氢化钠催化下与碳酸二乙酯发生羰基α-C-酰化反应生成3-氧代-3-(2-噻吩基)-丙酸乙酯75。酮酸酯75在手性催化剂存 在下用甲酸发生不对称还原反应生成(S)-3-羟基-3-(2-噻吩基)-丙酸乙酯74后与甲胺的甲醇溶液室温发生胺解反应制得(S)-N-甲基-3-羟基-3-(2-噻吩基)-丙酰氨73。醇 酰胺73用红铝或硼烷(由硼氢化钠和碘就地产生)将酰胺羰基还原为亚甲基生成(S)-3- N-甲氨基-1-(2-噻吩基)-1-丙醇72,然后用上述“合成路线一”类似的SNAr氧醚化反应方法,与1-氟萘反应制得度洛西汀64。
本信息由Chemicalbook晓楠编辑。盐酸度洛西汀(duloxetine hydrochloride)是新的选择性5-羟色胺和去甲肾上腺素再摄取双重作用的抑制剂,具有抗抑郁作用,同时对中枢性疼痛有抑制作用。药理学特点是能抑制神经元突触前膜对5-羟色胺和去甲肾上腺素的再摄取,对多巴胺再摄取的抑制作用较低。盐酸度洛西汀的适应症是抑郁症,它对内源性和非内源性抑郁、以及抑郁症伴发的疼痛症状都有效。治疗剂量为60mg/d~120mg/d,安全性好,不良反应少,较常见的不良反应有恶心、口干、便秘、食欲低下、疲劳、嗜睡和出汗增多。另一个适应证是糖尿病性神经病变所致的疼痛。
【商品名】 Cymbalta
【别 名】 LY-264453,LY-248686
【药效类别】 抗抑郁药,治疗紧张性尿失禁
【CAS】 136434-34-9
【研制单位】 日本Shionogi公司,美国Eli-Lilly公司
度洛西汀是一种选择性的5-HT与NE再摄取抑制剂(SSNRI)。度洛西汀抗抑郁与中枢镇痛作用的确切机制尚未明确,但认为与其增强中枢神经系统5-羟色胺与去甲肾上腺素能功能有关。临床前研究结果显示,度洛西汀是神经元5-HT与NE再摄取的强抑制剂。对多巴胺再摄取的抑制作用相对较弱。体外研究结果显示,度洛西汀与多巴胺能受体、肾上腺素能受体、胆碱能受体、组胺能受体、阿片受体、谷氨酸受体、GABA 受体无明显亲和力。度洛西汀不抑制单胺氧化酶。
图1为度洛西汀分子结构式。度洛西汀口服后,半衰期大约为12小时(变化范围为8~17小时),在治疗范围之内其药代动力学参数与剂量成正比,一般服药3天后达到稳态血药浓度。度洛西汀主要经肝脏代谢,涉及两种P450酶:CYP2D6和CYP1A2。
口服度洛西汀后,平均滞后2小时,药物开始吸收(Tlag);口服6小时后度洛西汀达到最大血浆浓度(Cmax),进食不影响Cmax,但是将延迟达峰时间6~10小时,略微降低吸收程度,约10%。晚间一次服药与晨间一次服药相比,度洛西汀的吸收滞后3小时,表现清除增加1/3。
表观分布容积平均为1640升。度洛西汀与人体血浆蛋白有高度亲和性(大于90%),主要与白蛋白和α1-酸性糖蛋白结合。 目前为评价度洛西汀和其他高蛋白结合药物之间是否有药物相 互作用。肝或肾功能不全不影响度洛西汀的血浆蛋白结合。
度洛西汀在人体内代谢广泛,代谢产物多。血浆中的代谢产物包括葡萄糖醛酸结合的4-羟基度洛西汀、硫酸结合的5-羟基-6-甲氧基度洛西汀。尿中分离出多种其他代谢产物,有些仅出现在小的消除代谢旁路中。尿液中仅有少量未经代谢的盐酸度洛西汀原型(1%),大部分(70%)以盐酸度洛西汀代谢产物形式经尿液排出,大约20%经粪便排出。用于治疗抑郁症。吞服,不要咀嚼和压碎。推荐起始剂量为40mg/d(40mg一日一次或20mg,一日两次)至60mg/d(一日一次),不考虑进食影响。1.禁用于已知对度洛西汀或产品中任何非活性成分过敏的患者。
2.禁止与单胺氧化酶抑制剂(MAOIs)联用,也不可以在MAOIs停药14天内使用本品;根据度洛西汀的半衰期,停用度洛西汀后至少5天,才能开始使用MAOIs。
3.临床显示度洛西汀有增加瞳孔散大的风险,因此未经控制的闭角型青光眼患者应避免使用度洛西汀。1.肝肾功能不全,会明显减低度洛西汀的代谢和清除,不建议用于该类患者。
2.度洛西汀与酒精交互作用可能会造成肝损伤,通常不建议用于治疗酗酒患者。
3.临床试验中,度洛西汀与安慰剂相比,收缩压平均升高2mmHg,舒张压平均升高0.5mmHg。建议在治疗前及治疗中定期测量血压。度洛西汀通过两种CYP2D6和CYP1A2代谢,中度抑制CYP2D6,但不抑制也不诱导CYP1A2和CYP3A4。与其他主要通过CYP2D6,且治疗窗狭窄的药物(如:TCAs、Ic类抗心律失常药物、吩噻嗪)时,应谨慎。【孕妇即哺乳期妇女用药】 妊娠分类C,怀孕妇女服用本品是否安全尚不明确。对于孕妇,应权衡利弊决定是否服用本品。只有当潜在利益高于风险才可以使用,否则怀孕期及哺乳期内不应使用。
【儿童用药】 目前尚缺乏在儿童中的足够临床经验。
【老年人用药】 临床研究中未观察到老年人群与年轻人群在安全性和疗效方面的显著差异,但不能排除某些老年患者的敏感性增高。度洛西汀的合成路线按照醚化反应所用原料的不同,主要可分为以下两大类:①以(S)-3-N,N-二甲氨基-1-(2-噻吩基)-1-丙醇67为原料;②以(S)-3-N-甲氨基-1-(2-噻吩基)-1-丙醇72为原料。
1. 以2-乙酰噻吩71为原料,在乙醇中与二甲胺盐酸盐69和多聚甲醛 70发生Mannich反应生成3-N,N-二甲氨基-1-(2-噻吩基)-1-丙酮68。酮叔胺68经硼氢化钠还原为醇叔胺67的消旋体,接着用(S)-(+)-扁桃酸进行拆分得到(S)-3-N,N- 二甲氨基-1-(2-噻吩基)-1-丙醇67。手性醇叔胺67在氢化钠作用下于二甲亚砜(DMSO)中与1-氟萘进行SNAr氧醚化反应,生成N-甲基度洛西汀65,再与氯甲酸苯酯(或氯甲酸三氯乙酯)反应生成度洛西汀的氨基甲酸酯88,最后不经分离直接水解就制得度洛西汀64。这是早期合成度洛西汀的一条最基本、技术相对成熟的合成路线,也比较适合于工业化生产。
图2为以2-乙酰噻吩为原料合成度洛西汀的路线图。
2. 以2-乙酰噻吩71为起始原料,在氢化钠催化下与碳酸二乙酯发生羰基α-C-酰化反应生成3-氧代-3-(2-噻吩基)-丙酸乙酯75。酮酸酯75在手性催化剂存 在下用甲酸发生不对称还原反应生成(S)-3-羟基-3-(2-噻吩基)-丙酸乙酯74后与甲胺的甲醇溶液室温发生胺解反应制得(S)-N-甲基-3-羟基-3-(2-噻吩基)-丙酰氨73。醇 酰胺73用红铝或硼烷(由硼氢化钠和碘就地产生)将酰胺羰基还原为亚甲基生成(S)-3- N-甲氨基-1-(2-噻吩基)-1-丙醇72,然后用上述“合成路线一”类似的SNAr氧醚化反应方法,与1-氟萘反应制得度洛西汀64。
用途
是一种5-羟色胺和去甲肾上腺素重摄取的双重抑制剂,能有效治疗抑郁症的情感和躯体症状安全信息
下游产品