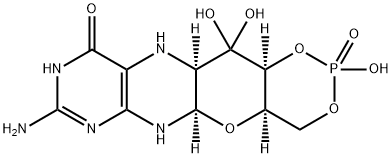
How is Fosdenopterin synthesised?
Jan 16,2024
Synthesis of Fosdenopterin
The synthesis of Fosdenopterin starts with d-galactose as the starting material and begins with the synthesis of the core structure, which is finally completed by vortex oxidation of secondary alcohols, formation of hydrates from the resulting ketones and subsequent global deprotection. The specific synthesis steps are as follows:
Step 1:Preparation of Fosdenopterin Core
Treatment of D-galactose (11.1) with phenylhydrazine gave galactose phenylhydrazone 11.2 in equilibrium with the tautomeric isomer 11.3 in 80% yield. This key starting material was used to rapidly enable synthesis of core structure 11.5 as a single isomer in 1 step via heating (110 °C) with 2,5,6-triamino-3,4- dihydropyrimidinone 11.4 in MeOH/water (36% yield).

Remarkably, this sequence established each stereocenter present in the final drug substance. This transformation is proposed to take place initially via an Amadori rearrangement of the phenylhydrazone (conversion of 11.2 to 11.3), followed by regioselective condensation of the resultant ketone with the most basic amine within 11.4 (the 5-NH2 group), and subsequent cyclization and pyran formation via intramolecular addition of the C5 hydroxyl group present in galactose. Further, while full mechanistic details are not provided, generation of a single diastereomer by this reaction sequence in similar systems has led to the hypothesis that the C5 hydroxyl group participates in the entire cyclization, leading exclusively to formation of the cis isomer at the ring junction.
Step 2:Preparation of Fosdenopterin
With advanced intermediate 11.5 in hand, protection of N10 was pursued as a means to prevent any undesired oxidation of 11.5 to its corresponding aromatic pterin derivative. Therefore, 11.5 was subjected to excess Boc anhydride (9 equiv) and DMAP in THF, leading to a mixture of products, each with varying levels of Boc incorporation, including intermediates 11.6 and 11.7 (hepta- and hexa-Boc protection, respectively). It was noted, however, that stability of 11.6 was limited, and simply subjecting 11.6 to silica gel furnished hexa-protected intermediate 11.7. Based on this, both 11.6 and 11.7 could be funneled to tri-Boc-amino-protected intermediate 11.8 by addition of carefully controlled quantities of NaOMe in MeOH, and subsequent workup with acidic Dowex resin provided the target compound in 75% overall yield. Treatment of 11.8 with methyl dichlorophosphate and pyridine in DCM provided the desired phosphate ester 11.9 in 65% yield.
The finalsteps of the synthesis were completed by Swern oxidation of the secondary alcohol, hydrate formation from the resultant ketone, and subsequent global deprotection. As the hydrate proved to be unstable after workup, this crude intermediate was immediately treated with bromotrimethylsilane in aqueous acetone, leading to complete amine and phosphonate ester deprotection, and yielding fosdenopterin after crystallization. Recrystallization of fosdenopterin from aqueous hydrobromic acid/2-propanol provided fosdenopterin monobromide dihydrate in 95% purity.

- Related articles
- Related Qustion
Sotagliflozin (LX-4211) is a dual sodium-glucose co-transporter-2 and 1 (SGLT2/1) inhibitor for the treatment of both type 1 (T1D) and type 2 diabetes (T2D).....
Jan 16,2024APIOdevixibat is synthesised in two steps by chemical reaction using enfumafungin as a raw material.....
Jan 16,2024Inhibitors