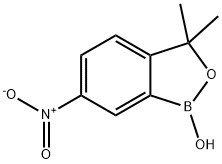
3,3-diMethyl-6-nitrobenzo[c][1,2]oxaborol-1(3H)-ol synthesis
- Product Name:3,3-diMethyl-6-nitrobenzo[c][1,2]oxaborol-1(3H)-ol
- CAS Number:1266084-47-2
- Molecular formula:C9H10BNO4
- Molecular Weight:206.99
![3,3-diMethyl-6-nitrobenzo[c][1,2]oxaborol-1(3H)-ol](/NewsImg/2023-10-17/6383313396958092738111157.jpg "3,3-diMethyl-6-nitrobenzo[c][1,2]oxaborol-1(3H)-ol")
![3,3-diMethylbenzo[c][1,2]oxaborol-1(3H)-ol](/CAS/GIF/221352-10-9.gif)
221352-10-9
76 suppliers
$40.00/100mg
1266084-47-2
19 suppliers
$45.00/2.5mg
Yield:1266084-47-2 89%
Reaction Conditions:
with nitric acid at -45; for 0.5 h;
Steps:
Preparation of benzoxaborole 2i
A mixture of methyl 2-bromobenzoate (95 g, 0.44 mol) in 400 mL of THF cooled to 5 °C and was added CH3MgCl (400 mL, 0.12 mol) dropwise below 10 °C under N2. The mixture was stirred at room temperature for 2h and quenched with 500 mL of sat. NH4Cl solution. The mixture was extracted with ethyl acetate and the combined organic layer was washed with brine, dried over sodium sulfate and concentrated under vacuum to provide 90 g of yellow oil. The crude product was used directly in the next without any further purification.NaH (22 g, 0.55 mol) was suspended in 600 mL of THF and 2-(2-bromophenyl)propan-2-ol (86 g, 0.4 mol) in 200 mL of THF was added dropwise at room temperature. After addition the mixture was heated to reflux for 2h. Then the mixture was allowed to cool to 0 °C and chloromethyl methyl ether (3 0 g, 0.38 mol) was added below 10 °C. The mixture was stirred at room temperature overnight followed by filtration. The filtrate was then concentrated under vacuum and the residue was used for column chromatography on silica gel to give the yellow oil (45 g, 43%).To a solution of 1-bromo-2-(2-(methoxymethoxy)propan-2-yl)benzene (10.4 g, 40 mmol) and B(iPrO)3 (8 g, 43 mmol) in 100 mL of THF at -78 °C under N2 was added dropwise n-BuLi (20 mL, 50 mmol). The mixture was stirred at -78 °C for 30 min and then it was allowed to warm to room temperature and continued to stir for 2h. After that it was quenched with 6 N HCl (20 mL) and the resulting mixture was concentrated under vacuum. The residue was re-dissolved in MeOH (80 mL) and 6 N HCl (40 mL) and heated at reflux for 2h. Then it was cooled to room temperature and diluted with water followed by extraction with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified on column chromatography on silica gel with the eluent ethyl acetate/petroleum ether = 1/2 to give a yellow oil (4 g, 62%). 1H NMR (300 MHz, DMSO-d6): δ 9.01 (s, 1 H), 7.65 (d, 1H), 7.31 (m, 3H), 1.49 (s, 6H).To a solution of 10 mL of HNO3 at -45 °C was added compound 3,3-dimethylbenzo[c][1,2]oxaborol-1(3H)-ol (700 mg, 4.32 mmol). The mixture was stirred at -45 °C for 30 min. Then it was warmed to room temperature and diluted with water followed by extraction with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated to provide a yellow oil (800 mg, 89%). 1H NMR (300 MHz, CDCl3): δ 8.56 (s, 1H), 8.35 (m, 1H), 7.42 (m, 1H), 1.59 (s, 6H).Compound 3,3-dimethyl-6-nitrobenzo[c][1,2]oxaborol-1(3H)-ol (7 g, 7.2 mmol) was dissolved in 30 mL of methanol and was added 3 mL of conc. HCl and Pd/C (1 g). The hydrogenation was conducted at room temperature under one atmosphere of hydrogen overnight. Then the mixture was filtered and the filtrate was concentrated to provide the product 2i as HCl salt form (1.4 g, 90%). LC-MS (ESI) m/z 178 [M+1]+, (calcd MS 177.1). 1H NMR (300 MHz, CD3OD): δ 7.54 (m, 3H), 1.59 (s, 6H).
References:
Li, Xianfeng;Zhang, Suoming;Zhang, Yong-Kang;Liu, Yang;Ding, Charles Z.;Zhou, Yasheen;Plattner, Jacob J.;Baker, Stephen J.;Bu, Wei;Liu, Liang;Kazmierski, Wieslaw M.;Duan, Maosheng;Grimes, Richard M.;Wright, Lois L.;Smith, Gary K.;Jarvest, Richard L.;Ji, Jing-Jing;Cooper, Joel P.;Tallant, Matthew D.;Crosby, Renae M.;Creech, Katrina;Ni, Zhi-Jie;Zou, Wuxin;Wright, Jon [Bioorganic and Medicinal Chemistry Letters,2011,vol. 21,# 7,p. 2048 - 2054] Location in patent:supporting information; experimental part

610-94-6
299 suppliers
$13.00/25g
1266084-47-2
19 suppliers
$45.00/2.5mg

7073-69-0
88 suppliers
$22.00/250mg
1266084-47-2
19 suppliers
$45.00/2.5mg
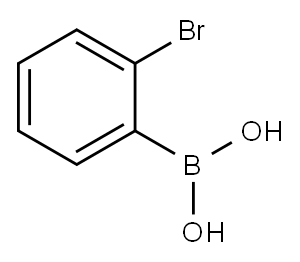
244205-40-1
324 suppliers
$9.00/1g
1266084-47-2
19 suppliers
$45.00/2.5mg
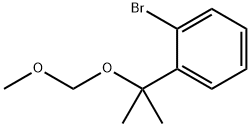
845302-03-6
3 suppliers
inquiry
1266084-47-2
19 suppliers
$45.00/2.5mg