유전적인 결함을 일으킬 것으로 의심됨 (노출되어도 생식세포 유전독성을 일으키지 않는다는 결정적인 증거가 있는 노출경로가 있다면 노출경로 기재)
생식세포 변이원성 물질
구분 2
경고
P201,P202, P281, P308+P313, P405,P501
H361
태아 또는 생식능력에 손상을 일으킬 것으로 의심됨
생식독성 물질
구분 2
경고
P201, P202, P281, P308+P313, P405,P501
H373
장기간 또는 반복 노출되면 장기(또는, 영향을 받은 알려진 모든 장기를 명시)에 손상을 일으킬 수 있음
특정 표적장기 독성 - 반복 노출
구분 2
경고
P260, P314, P501
예방조치문구:
P201
사용 전 취급 설명서를 확보하시오.
P202
모든 안전 조치 문구를 읽고 이해하기 전에는 취급하지 마시오.
P260
분진·흄·가스·미스트·증기·...·스프레이를 흡입하지 마시오.
P281
요구되는 개인 보호구를 착용하시오
P308+P313
노출 또는 접촉이 우려되면 의학적인 조치· 조언를 구하시오.
P314
불편함을 느끼면 의학적인 조치·조언을 구하시오.
P405
밀봉하여 저장하시오.
P501
...에 내용물 / 용기를 폐기 하시오.
Alectinib Hydrochloride C화학적 특성, 용도, 생산
개요
Alectinib hydrochloride, developed by Chugai Pharmaceutical/
Hoffman-La Roche under the trade name Alecensa®, was approved
in Japan in April 2014 for the treatment of anaplastic lymphoma
kinase (ALK) fusion-gene positive, unresectable, advanced, or
recurrent non-small cell lung cancer (NSCLC). The compound is
a highly selective second-generation ALK inhibitor, and while
alectinib currently remains a focus of further development in Europe
and the U.S., the compound has been granted orphan drug designation
in Japan after showing a 93.5% objective response rate in
phase II clinical trials. In addition to providing rapid treatment
response time in a majority of patients, trials showed a 76%
2-year progression-free survival rate. Since the initial approval
of crizotinib—the first ALK inhibitor indicated for treatment of ALKrearranged
NSCLC —patients treated with crizotinib have shown
remarkable improvement as compared to treatment with other
chemotherapeutic methods,21 although drug resistance has shown
to be a major side effect of this therapy. Preliminary preclinical
and clinical studies of alectinib have shown significant promise
for overcoming drug resistance developed with other ALK
inhibitors.
용도
CH5424802 Hydrochloride is a highly selective and potent anaplastic lymphoma kinase (ALK) inhibitor capable of blocking the resistant gatekeeper mutant, which results in reduced cell growth. Also is an intermediate of Alectinib (C183360), a highly selective and potent anaplastic lymphoma kinase (ALK) inhibitor capable of blocking the resistant gatekeeper mutant, which results in reduced cell growth.
Synthesis
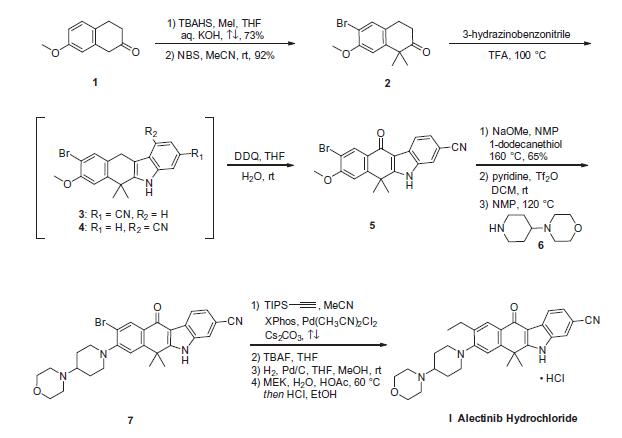
The synthetic route to alectinib as reported by Chugai
begins with 7-methoxy-2-tetralone (1). Bis-methylation
with tetrabutylammonium hydrogen sulfide (TBAHS)/aq KOH/MeI
followed by bromination with N-bromosuccinimide (NBS) provided
the bromo-tetralone 2 in 67% yield over the two steps. Further
reaction of 2 with 3-hydrazinobenzonitrile/trifluoroacetic acid (TFA) led to formation of the desired Fischer indole product,
albeit as a 1:1 mixture of regioisomers (3/4), which were carried
forward as a mixture to oxidation with 2,3-dichloro-5,6-dicyano-
1,4-benzoquinone (DDQ). It is important to note that although representative
procedures are published describing the conversion of
2 to alectinib (I), no yields were provided for these transformations.
Following oxidation, the desired product 5 could be isolated
as a single isomer via precipitation from the crude reaction mixture.
Installation of the 4-morpholino-piperidine moiety took place
in three transformations from 5, beginning with 1-dodecanethiol/
N-methyl-2-pyrrolidone (NMP)/NaOMe-facilitated methyl cleavage.
The corresponding phenol was then readily converted to the
triflate intermediate and displaced with 4-(piperidin-4-yl)morpholine
(6) at elevated temperature, providing intermediate 7. Crosscoupling
of the bromide 7 with ethynyl triisopropylsilane under
Pd-catalyzed cross-coupling conditions (Pd(CH3CN)2Cl2/2-dicyclohexylphosphino-
20,40,60-triisopropylbiphenyl (XPhos), reflux) followed
by cleavage of the resulting alkylsilane with
tetrabutylammonium fluoride (TBAF) yielded the ethynyl precursor
to alectinib. Hydrogenation of this unsaturated system under
standard conditions (H2, Pd/C) followed by HCl salt formation furnished
the final drug target alectinib hydrochloride (I).