近日,清华大学教授李隽教授和陕西理工大学于小虎副教授等人在Science China Materials上发表研究论文,基于性原理,系统研究了过渡金属单原子(铁,钴,镍,钌,铑,钯,锇,铱,铂)负载在磷钨酸(PTA)催化剂上的几何和电子结构,并进一步研究了乙烯环氧化在铁单原子催化剂上的可能反应机理。研究发现PTA最可能结合过渡金属单原子的位置是四配位中空位。乙烯环氧化催化活性的理论计算表明,非贵金属Fe1-PTA具有可观的吸附能,这是引发此催化循环的关键物理量。李教授等人进一步进行了铁单原子和PTA结合的成键分析,发现电荷从铁单原子转移到PTA团簇,并且强烈的Fe–O共价金属-载体相互作用(CMSI)是其高稳定性的基础。
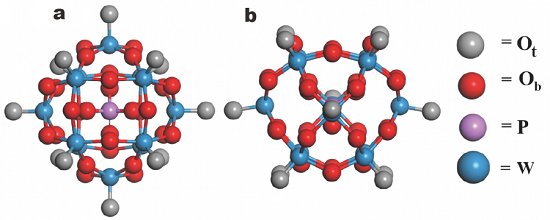
图1 PTA优化结构的俯视图和侧视图。
在催化剂Fe1-PTA上可能的乙烯环氧化催化机理共包括三步: 1)氧气分子通过电荷转移吸附在Fe1-PTA上;2)个乙烯分子攻击吸附在Fe1-PTA上的氧气分子,随后形成C2H4O;3)表面吸附的氧原子和第二个乙烯分子生成C2H4O,完成催化循环。本研究发现,Fe1-PTA对于乙烯环氧化主要通过Eley-Rideal 机理进行。
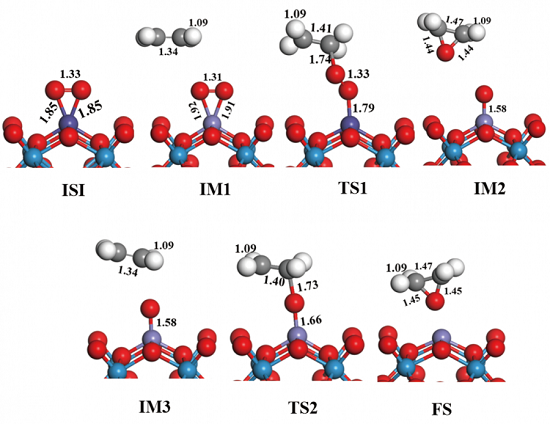
图2 Fe1-PTA上乙烯环氧化的初始态(IS),过渡态(TS)和终态(FS)的优化几何结构。
本工作为发展高效多相非贵金属单原子乙烯环氧化催化剂提供了理论依据。
该研究成果最近发表于Science China Materials, 2020, 10.1007/s40843-020-1399-y。