【背景及概述】[1][2]
氟康唑(fluconazole,FCZ)是一种可通过竞争性抑制真菌中麦角甾醇的合成从而抑制或杀灭真菌的三唑类抗真菌药物,对治疗深部真菌感染特别是白念珠菌及新型隐球菌有显著疗效。该药自1988年上市以来,因具有抗真菌谱广、肝毒性小、口服吸收好、生物利用度高、组织分布广等优良的药代动力学特性而在临床上广泛应用,被世界卫生组织WHO指定为治疗全身性真菌感染的首选药物。基于此,众多工作致力于对其深入研究与开发。随着真菌感染发生率的上升以及抗真菌药物在临床上的大量使用,真菌耐药性问题的解决迫在眉睫,仅凭研发新型抗真菌药物难以满足临床治疗的需求。
近年来,两种或两种以上药物的联合应用发迅速,成为临床抗真菌治疗的重要研究方向之一。从理论上讲,具有不同作用机制和作用位点的抗真菌药物的联合应用可能产生协同或相加效果,并可以减少单一用药的剂量及毒副作用,从而增强抗真菌活性及拓宽抗真菌谱。为了解决氟康唑目前出现的耐药性、不良反应及抗真菌谱窄等的问题,将其与其它抗真菌药物联合应用成为国内外研究的热点。目前氟康唑与其它药物联合应用的研究主要集中于以下5个方向:f1)与多烯类抗真菌药联用;(2)与丙烯胺类抗真菌药物联用;(3)与核酸抑制剂5一氟胞嘧啶联用;(4)与细胞壁抑制剂联用;(5)与其它药物联用。
【适应症】[3]
1. 念珠菌病用于治疗口咽部和食道感染;播撒性念珠菌病,包括腹膜炎、肺炎、尿路感染等;念珠菌外阴阴道炎。尚可用于骨髓移植患者接受细胞毒类药物或放射治疗时,预防念珠菌感染的发生。
2. 隐球菌病用于治疗脑膜炎以外的新形隐球菌病;在治疗隐球菌脑膜炎时,本品可作为两性霉素B联合氟胞嘧啶初治后的维持治疗药物。
3. 球胞子菌病。
4. 芽生菌病、组织胞浆菌病,本品可作为伊曲康唑的替代选用药物。
【规格】[3]
针剂:200mg∶100mL;片剂或胶囊剂:50mg,100mg,150mg,200mg。
【用法用量】[3]
脉给药,成人每日为100~ 200mg,小儿每日按体重3~ 6mg/kg;成人口服每日50~ 100mg,必要时150~ 300mg。
【药理作用】[4]
氟康唑为氟代三唑类抗真菌药,抗菌谱与酮康唑相似,抗菌活性比酮康唑强。其作用机制是抑制真菌细胞膜必要成分麦角甾醇合成酶,使麦角甾醇合成受阻,破坏真菌细胞壁的完整性,抑制其生长繁殖。本品对白色念珠菌、大小孢子菌、新型隐球菌、表皮癣菌及荚膜组织胞浆菌等均有强力抗菌活性。
【药代动力学】[3]
口服和静脉注射的药代动力学相似。口服吸收良好,血药浓度可达静脉滴注后90%以上,口服吸收不受食物影响。空腹服用本品后0.5~1.5小时血药浓度达高峰,血浆T1/2约为30小时。每日给药一次连续给药至第4~5日,血药浓度已达稳态浓度的90%,代谢80%以原形随尿排泄。本品能良好透入全身体液,在唾液和痰中的浓度接近血药浓度。在真菌性脑膜炎患者的脑脊液中的浓度约为血药浓度的80%。
【不良反应】[5]
主要有轻度消化道反应,其次为皮疹等过敏反应,少数患者可出现头痛、头晕、失眠等神经系统反应。也可有一过性血清转氨酶及血肌酐值升高。
【药物相互作用】[3]
1.与华法令合用,能延长凝血酶原时间,故应调整华法令的剂量。
2.本品可延长磺胺脲类降血糖药的血浆半衰期。
3. 与苯妥英钠合用,可增加苯妥英钠的血浓度,如需合用,应对苯妥英钠的血浓度进行监测。
4. 与利福平合用,氟康唑的血浓度降低,应增加氟康唑的剂量。
【注意事项】[3]
孕妇、哺乳期妇女和肝肾功能不良者慎用。
【制备】[6]
一种氟康唑的制备方法,其特征在于:以式(I)表示的氟康唑按照如下步骤获得:
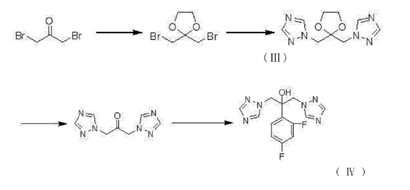
A.2,2‑二溴甲基‑1,3‑二氧戊烷(II)的制备
在反应器中加入1,3‑二溴丙酮1倍,甲苯4‑8倍(重量比),乙二醇0.31‑0.38倍 (重量比)和催化剂对甲基苯磺酸0.01‑0.03倍(重量比),升温至100℃搅拌反应,然后在此温度下回流分水,直至馏出液为清液,约6小时,然后冷却至室温,过滤除去不溶物,滤液升温至120℃蒸馏除去甲苯,得到的残余物为2,2‑二溴甲基‑1,3‑二氧戊烷粗品,减压蒸馏收集121‑124℃/10mmHg馏分,即2,2‑二溴甲基‑1,3‑二氧戊烷(II)。
B.1,1'‑(2,2‑双亚甲基‑双1H‑1,2,4‑三氮唑)‑1,3‑二氧戊烷(III)的制备
在反应器中加入二氯甲烷3‑6倍(重量比),在搅拌条件下加入1H‑1,2,4‑三氮唑0.53‑0.59倍(重量比),离子液体0.1倍(重量比)和碳酸钠0.81‑0.90倍或者碳酸钾1.06‑1.17倍(重量比),室温搅拌1小时,然后滴加2,2‑二溴甲基‑1,3‑二氧戊烷(II)1倍,滴加过程中反应混合物不断搅拌,滴加完毕后升温至35℃继续搅拌反应10‑20小时,然后冷却至室温,过滤除去不溶物,有机层用饱和食盐水洗涤后用无水硫酸钠干燥,过滤除去干燥剂无水硫酸钠后,滤液升温至50℃除去溶剂,得到的残余物为1,1'‑(2,2‑双亚甲基‑双1H‑1,2,4‑三氮唑)‑1,3‑二氧戊烷(III)粗品,该步产物不需要进一步纯化可直接用于下一步反应。此步骤中涉及的离子液体指的是3‑甲基‑1‑乙基咪唑硫酸氢盐,1,3‑二乙基咪唑硫酸氢盐和3‑丁基‑1‑乙基咪唑硫酸氢盐中间的一种。
C.1,3‑二(1H‑1,2,4‑三氮唑‑1‑基)丙酮(IV)的制备
在反应器中加入1,1'‑(2,2‑双亚甲基‑双1H‑1,2,4‑三氮唑)‑1,3‑二氧戊烷(III)1倍,浓度为20%或者浓度为25%的盐酸溶液4‑6倍(重量比),搅拌均匀后加热至100℃反应24‑36小时,反应结束后反应混合物冷却至室温,过滤除去不溶物,滤液用5%的氢氧化钠水溶液调节pH至12,然后混合液用二氯甲烷5倍(重量比)萃取2次,合并有机层后用无水硫酸钠干燥,过滤除去干燥剂无水硫酸钠后,滤液升温至50℃除去溶剂,得到1,3‑二(1H‑1,2,4‑三氮唑‑1‑基)丙酮(IV)粗品,经乙醇3倍(重量比)重结晶后得到精品1,3‑二(1H‑1,2,4‑三氮唑‑1‑基)丙酮(IV)。
D.氟康唑(I)的制备
在反应器中加入3,5‑二氟溴苯1.10‑1.21倍(重量比),无水四氢呋喃3‑4倍(重量比),镁屑0.13‑0.15倍(重量比)和单质碘0.05倍(重量比),加热至回流,引发反应后(单质碘颜色褪去)调节加热装置使混合物保持微沸状态,直至镁屑基本反应完全,约4小时,制备好的3,5‑二氟溴苯格式试剂待用。在另一反应器中加入1,3‑二(1H‑1,2,4‑三氮唑‑1‑基)丙酮(IV)1倍,无水四氢呋喃2‑3倍(重量比),搅拌均匀后滴加上述制备好的3,5‑二氟溴苯格式试剂,滴加过程保持混合物温度不超过50℃,滴加完毕后继续室温搅拌反应8‑10小时,反应结束,加入饱和氯化铵水溶液淬灭反应,分液,水相用乙酸乙酯3倍(重量比)萃取2次,合并有机相,后用无水硫酸钠干燥,过滤除去干燥剂无水硫酸钠后,滤液升温至80℃除去溶剂,得到氟康唑(I)粗品,粗品用75%的异丙醇水溶液3倍(重量比)重结晶,得到精品氟康唑(I)。
【主要参考资料】
[1] 米佳丽, 周成合, 白雪. 含三唑的抗微生物药物研究进展[J]. 中国抗生素杂志, 2007, 32(10): 587-593.
[2] 万昆, 张奕奕, 周成合, 等. 抗真菌药物氟康唑研究新进展[J]. 中国抗生素杂志, 2012 (2012 年 01): 8-15+ 20.
[3] 口腔临床药物手册
[4] 中国药房调剂
[5] 全科医生药物手册
[6] 宋苗根;王金银.一种氟康唑的制备方法. CN201010246142.8 ,申请日2010-08-05