背景及概述[1]
头孢他啶化学名称为(6R,7R)-7-[[(2Z)-2-(2-氨基-1,3-噻唑-4-基)-2-(1-羟基-2-甲基-1-氧代丙烷-2-基)氧亚氨乙酰]氨基]-8-氧代-3-(吡啶-1-鎓-1-基甲基)-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸。为第三代注射用头孢抗生素产品。根据EP药典收纳的头孢他啶甲酯(H)的结构式分析,在头孢他啶的制备过程中最有可能产生该杂质是在合成头孢他啶叔丁酯工艺环节或头孢他啶活性酯原料中有微量该杂质。通过相关研究文献确证,无论在他啶活性酯合成中还是他啶叔丁酯合成过程中都要控制溶媒甲醇的使用。
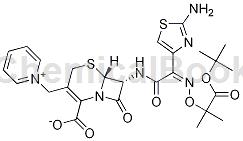
头孢他啶杂质H的结构为(6R,7R)-7-[[(2-氨基-1,3,4-噻唑-4-基)-2-(1-甲氧基2-甲基-1-氧代内烷-2-基)氧亚氨乙酰]氨基]-8-氧代-3-(吡啶-1-鎓-1-基-甲基)-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸;该杂质通过斑马鱼胚胎毒性实验,发现致畸作用是头孢他啶的25倍、致死作用是头孢他啶的8倍。所以头孢他啶杂质H应予以严格控制。目前国内合成头孢他啶主要是7-氨基头孢烷酸与硅烷化试剂在二氯甲烷溶媒中回流先进行氨基羧基保护,然后与三甲基碘硅烷形成碘代物再与吡啶合成7-APCA三位中间体。
7-APCA在二氯甲烷/甲醇等有机溶媒中与他啶活性酯碱性条件下缩合成他啶叔丁酯,他啶叔丁酯在甲酸/盐酸混合液中酸化形成他啶盐酸盐,他啶盐酸盐在适量注射用水中用稀碱调PH溶解,然后酸化析晶得到目标产物头孢他啶。上述过程,产生头孢他啶甲酯(H)的过程就是在头孢他啶叔丁酯合成中溶媒甲醇的使用导致七位侧链中的叔丁基与甲醇发生酯交换反应生成。
制备[1]
一种头孢他啶叔丁酯的制备方法:在洁净干燥的四口反应瓶中加入三氯甲烷520ml,加入叔丁醇30ml,降温至0-5℃。加入7-APCA70g,他啶活性酯105g,控制温度滴加三乙胺,调PH至7.0-9.0。控温0-10℃反应20-24小时。反应毕,加入纯水150ml,搅拌1-2小时,分出三氯甲烷。药液中滴入异丙醇析出头孢他啶叔丁酯,0-10℃搅拌养晶2小时。抽滤,用异丙醇洗涤,烘干后得头孢他啶叔丁酯干品。重量产率:145%,纯度98.9%,
主要参考资料
[1] CN201910160018.0一种降低头孢他啶杂质H的工艺方法