- Cefuroxime
-

- $0.00 / 1Kg/Bag
-
2024-11-02
- CAS:55268-75-2
- Min. Order: 1KG
- Purity: 98%min
- Supply Ability: 500kg
- Cefuroxime axetil
-
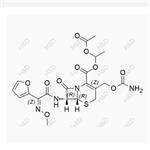
- $0.00 / 10mg
-
2024-09-20
- CAS:
- Min. Order: 10mg
- Purity: 98%
- Supply Ability: 500mg
- Cefuroxime
-

- $5.00 / 25kg
-
2024-04-24
- CAS:55268-75-2
- Min. Order: 1kg
- Purity: 99.92%
- Supply Ability: 50000tons
|
| | Cefuroxime Basic information |
| Product Name: | Cefuroxime | | Synonyms: | ethyl)-7-((2-furanyl(methyoxyimino)acetyl)amino)-8-oxo-,(6r-(6-alpha,7-beta(z;1-ACETYLOXYETHYL 4-(CARBAMOYLOXYMETHYL)-8-[2-(2-FURYL)-2-METHOXYIMINO-ACETYL]AMINO-7-OXO-2-THIA-6-AZABICYCLO[4.2.0]OCT-4-ENE-5-CARBOXYLATE;(6r-(6alpha,7beta(z)))-3-(((aminocarbonyl)oxy)methyl)-7-((2-furanyl(methoxyimino)a cetyl)-amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 1-(acetyloxy)ethyl ester;Cefuroxime Axetil EP 8.0 Impurity D;cefaloxime;-7-((Z);-3-((Carbamoyloxy);-2-(furan-2-yl) | | CAS: | 55268-75-2 | | MF: | C16H16N4O8S | | MW: | 424.39 | | EINECS: | 259-560-1 | | Product Categories: | Antibiotics;BacteriostaticAntibiotics;beta-Lactam StructureAlphabetic;C;CA - CG;Chemical Structure;Principle | | Mol File: | 55268-75-2.mol | 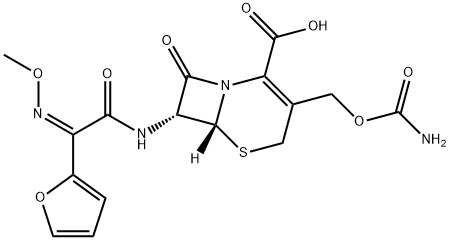 |
| | Cefuroxime Chemical Properties |
| Hazard Codes | Xn | | Risk Statements | 42/43 | | Safety Statements | 22-24-37-45 | | WGK Germany | 3 | | RTECS | XI0329000 |
| | Cefuroxime Usage And Synthesis |
| Description | Cefuroxime axetil is the acetoxyethyl ester and oral prodrug of cefuroxime, a
second-generation cephalosporin. It has a broad spectrum of action and is resistant to
most P-lactamases. Cefuroxime axed is indicated for serious bacterial infections,
especially where no identification of organism(s) has been made. | | Chemical Properties | white crystalline solid | | Originator | Glaxo (United Kingdom) | | Uses | Antibacterial. | | Uses | Cefuroxime is a second-generation cephalosporin antibiotic. | | Definition | ChEBI: A 3-(carbamoyloxymethyl)cephalosporin compound having a 7-(2Z)-2-(furan-2-yl)-2-(methoxyimino)acetamido side chain. | | Application | Cefuroxime was synthesized by Glaxo Laboratories in 1975 as the first cephem antibiotic
with the methoxyimino group at the 7 position. It is highly resistant to hydrolysis
by cephalosporinase and is active against a variety of gram-negative bacteria, including indolepositive Proteus, Enterobacter, and Citrobacter.
Cefuroxime is considered to be one of the socalled second-generation cephalosporins. | | Manufacturing Process | A stirred mixture of N,N-dimethylacetamide (75 ml), acetonitrile (75 ml),
triethylamine (42 ml, 0.3 mol) and (6R,7R)-7-amino-3-carbamoyloxy-methylceph-3-em-4-carboxylic acid was immersed in an ice-bath and water
(10 ml) was added. The mixture was stirred at 0°C to 2°C for 45 minutes, the
solid slowly dissolving to give a yellow solution.
Meanwhile a stirred suspension of phosphorus pentachloride (14.99 g, 0.072
mol) in dry dichloromethane (150 ml) was cooled to 0°C, and N,N-dimethylacetamide (27.5 ml) was added. The resulting solution was recooled
to -10°C and 2-(fur-2-yl)-2-methoxyiminoacetic acid (synisomer) (12.17 g,
0.072 mol) was added. The mixture was stirred at -10°C for 15 minutes and
crushed ice (35 g) was added. The mixture was stirred at 0°C for 10minutes,
where after the lower dichloromethane phase was added over 10 minutes to
the cephalosporin solution prepared above, cooled to -10°C so that the
reaction temperature rose steadily to 0°C. The mixture was stirred at 0°C to
2°C for 1 hour, where after the cooling bath was removed and the reaction
temperature allowed to rise to 20°C over 1 hour. The reaction mixture was
then added slowly to 2 N hydrochloric acid (100 ml) diluted with cold water
(1.15 l) at 5°C. The pH of the two phase mixture was adjusted to below 2
with 2 N hydrochloric acid (10 ml), and the mixture was stirred and recooled
to 5°C. The solid which precipitated was filtered, washed with
dichloromethane (100 ml) and water (250 ml), and dried in vacuo at 40°C
overnight to give the title compound (22.04 g, 86.6%). | | Brand name | Zinnat | | Therapeutic Function | Antibiotic | | Antimicrobial activity | The methoximino side chain provides stability
to most Gram-negative β-lactamases and it is active against
most enterobacteria, including many multiresistant strains.
Acinetobacter spp., S. marcescens and Ps. aeruginosa are resistant,
although some Burkholderia cepacia strains are susceptible.
Some anaerobic Gram-negative rods are susceptible, but
B. fragilis is resistant. The minimum immobilizing concentration
for the Nichol’s strain of T. pallidum is 0.01 mg/L. | | Pharmacology | Cefuroxime acts bactericidally. It has a narrow spectrum of antimicrobial action. It is resist�ant to beta-lactamase action. It is highly active with respect to Gram-negative microorgan�isms (intestinal and hemophilial bacilli, salmonella, shigella, enterobacteria, and gonococci).
It is also active with respect to Gram-positive microorganisms (staphylococci, streptococci).
It is inactive with respect to various types of Pseudomonas, most strains of enterococci, many
strains of Enterobacter cloacae, methylcillin-resistant staphylococci, and L. monocytogenes.
It is used for bacterial infections caused by microorganisms that are sensitive to the drug.
These may be abdominal and gynecological infections, sepsis, meningitis, endocarditis, infec�tions of the urinary and respiratory tracts, bones, joints, skin, and soft tissues. It is widely used
for pneumonia as well as bacterial meningitis in children, and for post-operational infectious
complications. Synonyms of this drug are ceftin, zinacef, curoxim, kefox, and many others. | | Pharmacokinetics | Oral absorption (axetil): 40–50%
Cmax 500 mg intramuscular: c. 18–25 mg/L after 0.5–1 h
0.75 g intravenous infusion: c. 50 mg/L end infusion
500 mg oral (axetil): 6–9 mg/L after 1.8–2.5 h
Plasma half-life: 1.1–1.4 h
Volume of distribution: 11–15 L
Plasma protein binding: 30%
Absorption
The acetoxyethyl ester (cefuroxime axetil) is rapidly hydrolyzed on passage through the intestinal mucosa and in the portal circulation to liberate cefuroxime, acetaldehyde and acetic acid. No unchanged ester is detectable in the systemic circulation. Absorption is independent of dose in the range 0.25–1 g, and there is no accumulation on repeated dosing. Bioavailability is improved after food to around 50%. In elderly subjects receiving doses of 500 mg every 8–12 h, peak plasma levels were 5.5 mg/L after 1.5–2 h in the fasting state, rising to 7.6 mg/L after 20 min when the dose was administered with food.
Distribution
In patients with severe meningeal inflammation, the mean CSF concentration after a 1.5 g intravenous dose was in the range 1.5–3.7 mg/L. In about one-third of patients with normal CSF, no drug could be detected and in the remainder concentrations were 0.2–1 mg/L. In children treated for meningitis with 50 or 75 mg/kg, the CSF:serum ratios were 0.07 and 0.10, respectively. Concentrations in pleural drain fluid after thoracic surgery approximated to serum levels at 2 h after doses of 1 or 1.5 g and exceeded serum levels at 4 h, when they were still around 10 mg/L. Levels in pericardial fluid were similar, with fluid:serum ratios of 0.44 between 0.5 and 2 h. In patients receiving 1.5 g by intravenous bolus preoperatively, concentrations around 22 mg/g were found in subcutaneous tissue at about 5 h with an elimination half-life of about 1.5 h.
Mean bone:serum ratios in the femoral head after 750 mg intramuscular and 1.5 g intravenous bolus injections were 0.14 and 0.23, respectively. In patients with chronic otitis media treated with 0.75 g every 8 h for 6–8 days, peak concentrations in the middle ear of 0.7–1.7 mg/L were reached about 2 h after the dose. In patients given 750 mg intramuscularly on five consecutive days the mean sputum concentration rose from 0.57 mg/L on the first day to 1.15 mg/L on the third.
Excretion
The drug is excreted unchanged in the urine mostly within 6 h of administration, producing concentrations exceeding 1 g/L. About 45–55% of the drug is excreted by tubular secretion, so that the administration of probenecid increases the serum peak and prolongs the plasma half-life. Renal clearance is slightly affected by the route of administration but lies between 95 and 180 L/min. The plasma half-life is prolonged in the elderly up to 2.4 h.
| | Clinical Use | It has been used successfully to treat urinary, soft-tissue
and pulmonary infections, as well as septicemia, and as a
single-
dose treatment (with probenecid) of gonorrhea due
to β-lactamase-producing strains. It has been widely used for
surgical prophylaxis. | | Clinical Use | Cefuroxime axetil (Ceftin) is the 1-acetyoxyethyl ester ofcefuroxime. During absorption, this acid-stable, lipophilic,oral prodrug derivative of cefuroxime is hydrolyzed to cefuroximeby intestinal and/or plasma enzymes. The axetilester provides an oral bioavailability of 35% to 50% of cefuroxime,depending on conditions. Oral absorption of theester is increased by food but decreased by antacids and histamineH2-antagonists. The latter effect may be because ofspontaneous hydrolysis of the ester in the intestine becauseof the higher pH created by these drugs. Axetil is used forthe oral treatment of non–life-threatening infections causedby bacteria that are susceptible to cefuroxime. The prodrugform permits twice-a-day dosing for such infections. | | Side effects | It is well tolerated with little pain or phlebitis on injection.
Minor hypersensitivity reactions and biochemical changes
common to cephalosporins are described.
The axetil ester may cause diarrhea and, in some cases,
vomiting. Changes in the bowel flora, sometimes with the
appearance of C. difficile, have been reported in about 15% of
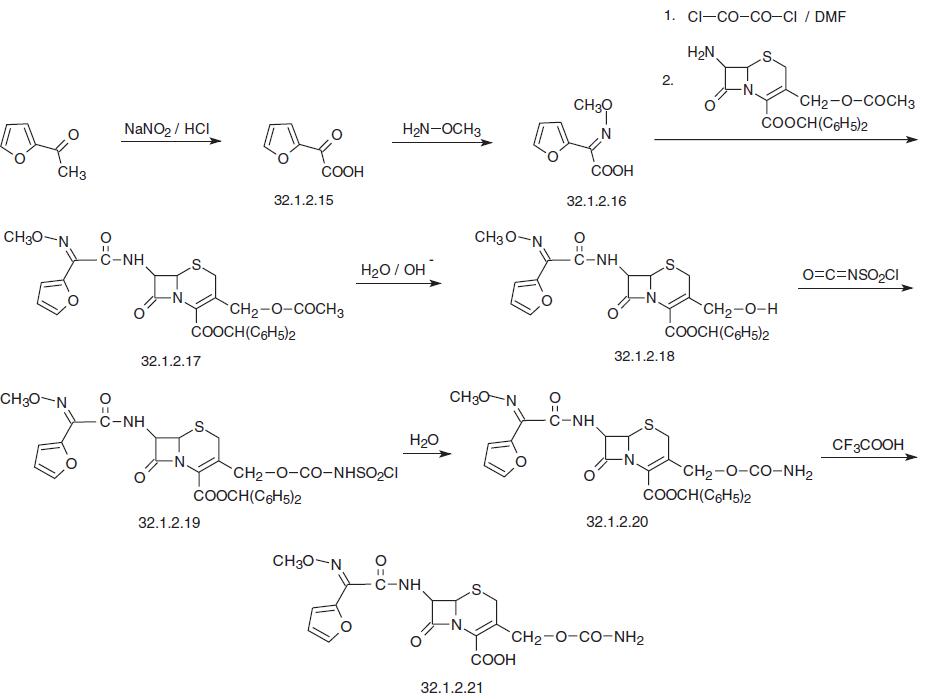
patients. Vaginitis is reported in about 2% of female patients. | | Synthesis | Cefuroxime, (Z)-mono(O-methyloxim) (6R,7R)-7-[2-(2-furyl)glyoxylamido]- 3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid carbamate (32.1.2.18), is synthesized from 2-acetylfuran. Oxidizing this compound with nitrous acid gives 2-furylglyoxalic acid (32.1.2.15), which is reacted with methoxylamine to give the corresponding oxime, syn-2-methoxyamino-2-(2-furyl)acetic acid (32.1.2.16), which is then transformed into a mixed anhydride when reacted with oxaloyl chloride in diemethylfor�mamide, and then reacted with benzhydryl ester of 7-aminocephalosporanic acid. The resulting product (32.1.2.17) undergoes enzymatic hydrolysis in an alkaline medium, in which the benzhydryl protection is not affected, and only the acetoxy group of the molecule at position C3 of the aminocephalosporanic acid is hydrolyzed. The resulting product with a free hydroxymethyl group (32.1.2.18) is reacted with chlorosulfonyl isocyanate, with intermediate formation of the corresponding N-chlorosulfonyl urethane (32.1.2.19), which is hydrolyzed by water to the urethane (32.1.2.20). Finally, removal of the benzhydryl protection using trifluo�roacetic acid gives the desired cefuroxime (32.1.2.21).
Another simpler way of the synthesis of cefuroxime is by direct acylation of 7-amino- 3-aminocarbonyloxymethyl-3-cefem-4-carboxylic acid (32.1.2.22), which is isolated from the cultural fluid of Streptomyces lactamdurans, using syn-2-methoxyamino-2- (2-furyl)acetic acid chloride, which is synthesized by reacting the corresponding acid with phosphorous pentachloride.
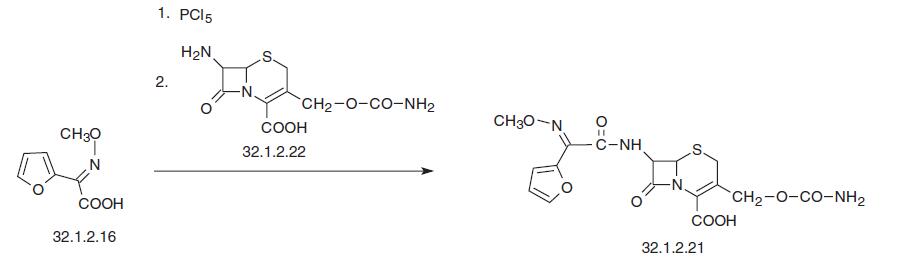 | | Veterinary Drugs and Treatments | Cefuroxime is a semi-synthetic 2nd generation cephalosporin with
enhanced activity against some gram-negative pathogens when
compared to the first generation agents. Cefuroxime is available in
both oral and parenteral dosage forms. It potentially may be useful
in small animals when a cephalosporin is desired to treat bacterial
infections susceptible to cefuroxime, but resistant to first generation
cephalosporins, when enhanced gram-negative coverage is desired
for surgery prophylaxis, or when high CNS levels are necessary.
Little information is available with regard to its clinical use in small
animals, however. |
| | Cefuroxime Preparation Products And Raw materials |
|