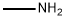Yield:-
Reaction Conditions:
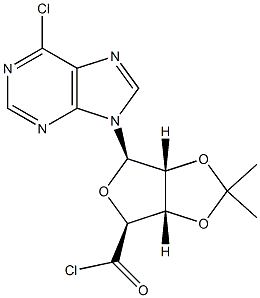
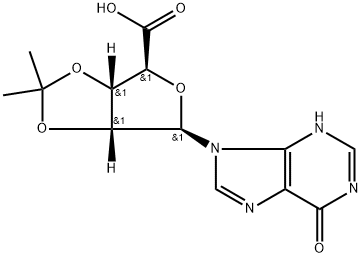
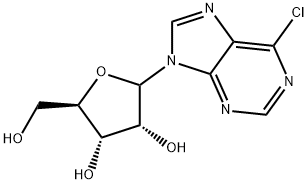
Stage #1: (3AS,4S,6R,6AR)-6-(6-chloro-purin-9-yl)-2,2-dimethyl-tetrahydro-furo[3,4-d][1,3]dioxole-4-carboxylic acidwith thionyl chloride in acetonitrile at 30; for 4 h;
Stage #2: methylaminewith N-ethyl-N,N-diisopropylamine in tetrahydrofuran;acetonitrile at 7; for 13 h;
Steps:
3
To a 72-L reactor, was charged 6-chloropurine acid [2600 g, 7.63 mol, 1.0 wt=1.0 vol] using CH3CN (31.7 L, 12.2 vol) to effect the transfer, and stirring commenced. Thionyl chloride (889 mL, 12.2 mol, 1.60 equiv, 0.342 vol) was added to the thick gray slurry and the mixture stirred at <30°C for 4h. Approximately 0.5 mL of the resultant dark solution was added to MeOH (2 mL, HPLC grade, Fisher), and analysis by TLC (UV detection, IPAc/MeOH, 10:1) showed the disappearance of the starting material. Over a period of 6.5 h, the batch solution was concentrated under vacuum on the rotary evaporator until distillation ceased (the water bath was initially set at 25 + 5 °C and gradually increased to 35 + 5 0C for this); CH3CN (825 mL, 0.32 vol) was used to rinse the reactor. The water-bath heat source was switched off and CH3CN (7.6 L, 2.9 vol) added to the residue in the bulb. Without vacuum, the bulb was rotated until the batch became fully mobile and then it was transferred to a 72-L reactor positioned in a steel tub. Stirring was commenced and additional CH3CN (25.5 L, 9.8 vol) was added. The batch was cooled using an ice-water/solvent bath until the internal batch temperature was <2 0C (this took 1 h); then 2 M methylamine in THF (4004 mL, 8.01 mol, 1.05 equiv, 1.54 vol) was added via a 5-L addition funnel over a period of 52 min while maintaining the internal batch temperature <7 0C. Subsequently, DIPEA (1976 mL, 11.3 mol, 1.5 equiv, 0.76 vol) was added via a 5-L addition funnel over 1 h; stirring was continued at <7 0C for a minimum of 1 h, the cooling bath was drained, and stirring continued for 11 h while the batch was allowed to warm to ambient temperature (the pH of the batch was 9). Typically the minimum temperature of the batch for these operations was 0 0C. Analysis by TLC (UV detection, IPAc/MeOH, 10:1) showed the disappearance of the 6- chloropurine acid/acyl chloride and the formation of one higher-running major product. The batch was transferred to another 72-L reactor setup in a heating mantle, equipped with a water-cooled condenser; CH3CN (1.3 L, 0.5 vol) was used to aid with the transfer. Stirring was commenced and 3-iodobenzylamine*HCl (2777 g, 10.30 mol, 1.35 equiv, 1.068 wt) was added. Subsequently, DIPEA (6656 mL, 38 mol, 5.0 equiv, 2.56 vol) was added and the mixture heated at 70 + 5 0C for 25 h. Analysis by HPLC showed 0.70% of the 6-chloropurine amide remaining by conversion, thus just meeting the <0.70% specification. The heat source was switched off and the batch allowed to cool overnight. Over a period of 8 h, the batch was concentrated under vacuum on the rotary evaporator at 40 +/- 5 0C until distillation ceased; CH3CN (1650 mL, 0.63 vol) was used to rinse the reactor. With the aid of IPAc (5.36 L, 2.1 vol), the residue was transferred to a 72-L reactor. Additional IPAc (20.4 L, 7.85 vol) was added to the reactor and stirring initiated. To this was added saturated aqueous sodium bicarbonate (22.2 L, 8.5 vol). After 30 min of stirring, the biphasic system was allowed to settle for 10 min and the lower aqueous phase collected (additional water (5 L) was added to dissolve the minor amount of remaining solids in the biphasic mixture). The remaining organic phase was washed with water (12.7 L, 4.9 vol) with 25-min stirring time and 38-min settling time. The combined organic phase was concentrated under vacuum on the rotary evaporator at 40 +/- 5 0C until distillation ceased (over a period of 6 h). The carboy was rinsed with IPAc (825 mL, 0.32 vol). In stages the resulting residue was slurried in the rotary evaporator bulb with MeOH (12.7 L, 4.9 vol) and concentrated until distillation ceased (4.5 h). This afforded crude IB-MECA acetonide (7.2 kg) as a damp beige solid which was stored for further processing and batch combination. Analysis by HPLC showed IB-MECA acetonide at 91.3 area % purity.Purification of Crude IB-MECA AcetonideThe crude MeOH damp IB-MECA acetonide (15 kg, 91 area %) was recrystallized from MeOH (34 L) at 60-65 °C. The resulting filter cake was rinsed with chilled MeOH (15.3 L) and transferred to eight drying trays (batch weight 10 kg) Analysis by HPLC showed IB-MECA acetonide at 65 area % purity present in the filtrate. It was estimated from the peak height that 1 kg of this material was sacrificed to this operation. The batch was dried in the vacuum oven set at 40 0C for approximately 22 h (7945 g). Analysis by HPLC showed the desired product at 99.0 area % purity contaminated with two significant impurities at RRT 0.64 (0.32 area %) and RRT 1.30 (0.58 area %). The mass spectra of these peaks are:RRT= 0.64MW = 424.19 m/e: 424.19 (100%), 425.19 (23.9%), 426.19 (4.0%), 425.18 (2.2%)C, 59.42; H, 5.70; N, 19.80; O, 15.08.RRT 1.30MW=752.34 gr/mol m/e: 752.01 (100.0%), 753.01 (32.4%), 754.02 (4.5%), 754.01 (1.5%) C, 43.10; H, 3.48; I, 33.74; N 11.17; O 8.51This material was recrystallized again from MeOH (23.8 L) at 60-65 °C. The resulting filter cake was rinsed with chilled MeOH (2 x 8 L). The total filtration time was 85 minutes. The cake was stored under a flow of N2. Analysis of the cake by HPLC showed the desired product at 99.77 area % purity contaminated with two significant minor impurities at RRT 0.64 (0.07 area %) and RRT 1.30 (0.16 area %). The batch was transferred to six drying trays (batch weight 7.4 kg) rather than perform the third optional recrystallization. The batch was dried in the vacuum oven set at 40 0C for approximately 6Oh. This afforded IB-MECA acetonide (7097 g) as a white solid after packaging into four amber glass jars; storage was at ambient temperature.
References:
WO2008/111082,2008,A1 Location in patent:Page/Page column 31-34
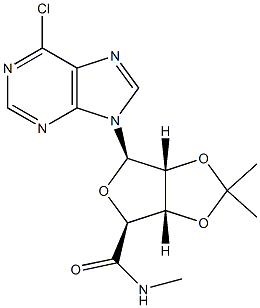
![(3AS,4S,6R,6AR)-6-(6-chloro-purin-9-yl)-2,2-dimethyl-tetrahydro-furo[3,4-d][1,3]dioxole-4-carboxylic acid](/CAS/20180703/GIF/120355-42-2.gif)