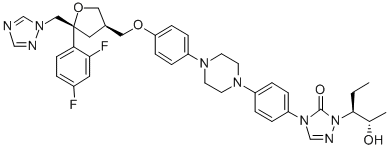
Posaconazole synthesis
- Product Name:Posaconazole
- CAS Number:171228-49-2
- Molecular formula:C37H42F2N8O4
- Molecular Weight:700.78
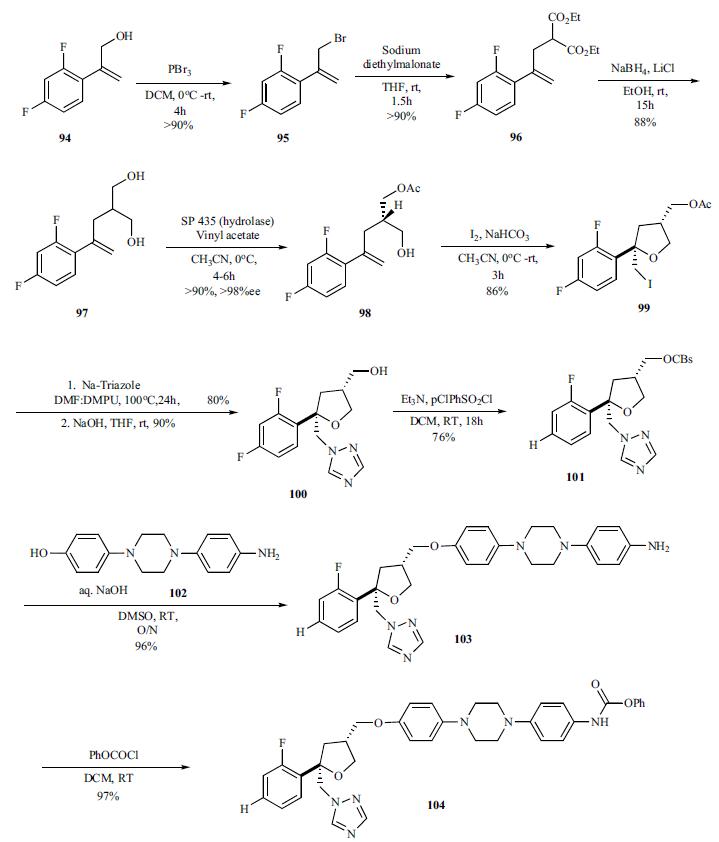
For the preparation of chiral hydrazine 107, intermediate needed to make the triazolone, lactam 105 was reduced with Red-Al to give (S)-2-benzyloxy propanal 106 (94%) which was then reacted with formyl hydrazine to give hydrazone 107 in 81% yield. Addition of EtMgBr directly to formyl hydrozones 107 gave mixture of (S,S)stereoisomer 109 and (S,R)-diastereomer 110 in relative good diastereoselectivity (94:6) in 55% yield. However, protection of the formyl group as TBDMS ether 108 followed by treatment of the EtMgCl gave 95% yield of the (S,S)-diastereomer 109 and (S,R)-diastereomer 110 in 99:1 ratio.
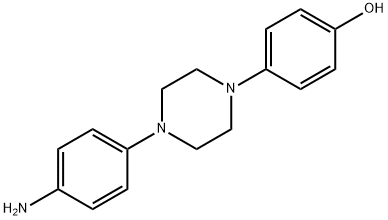
For finishing off the synthesis, the formyl hydrazine 109 was coupled with the phenyl carbamate 104 in toluene at 75 - 85°C for 12 – 24 hrs. After the completion of coupling, the intermediate was heated at 100 – 110°C for 24 – 48 hrs to completely cyclize to the benzyloxy triazolone 108, which was deprotected with 5% Pd/C and formic acid at room temperature overnight and 40°C for 24 h to give posaconazole (XV) in 80% overall yield.

170985-86-1
45 suppliers
$170.00/1mg
171228-49-2
645 suppliers
$6.00/5mg
Yield:171228-49-2 95%
Reaction Conditions:
with hydrogenchloride;water at 25 - 63;Temperature;
Steps:
7 Example -7
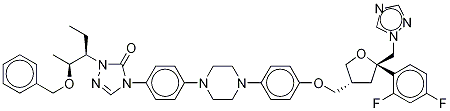
Example -7: Preparation of 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro- 5(lH-l,2,4-triazol-l-ylmethyl)-3-furanyl]methoxy]phenyl]-l-piperazinyl]phenyl]-2- [(lS,2S)-l-ethyl-2-hydroxypropyl]-2,4-dihydro-3H-l,2,4-triazol-3-one of the structural formula (IV) of amorphous form. 4-(4-(4-(4-(((3R,5R)-5-((lH-l,2,4-triazol-l-yl)methyl)-5-(2,4- difluorophenyl)tetrahydrofuran -3-yl)methoxy)phenyl)piperazin- 1 -yl)phenyl)- 1 -((2S,3S)- 2- (benzyloxy)pentan-3-yl)-lH-l,2,4-triazol-5(4H)-one of the structural formula (III) (350.0 g) of crystalline form B-3 was charged to a RBF containing Concentrated Hydrochloric Acid (5.0 vol.) at 25±2°C. The reaction mixture was slowly heated to 63+2 °C and stirred at 63±2°C for 2 to 3h. Reaction mass was cooled to 25±2°C and Dichloromethane (5.0 vol. X 2 times) was charged, stirred and layers were separated. The aqueous layer was added dichloromethane (6.0 vol.) and water (5.0 vol.). The contents were cooled to 15±5°C, pH of the above mass was adjusted to 10 to 12 pH using 25% aqueous sodium hydroxide. Layers were separated. The aqueous layer was re-extracted with dichloromethane (5.0 vol. X 2 times). Combined dichloromethane layers were washed with 10 % aqueous sodium hydroxide (3.0 vol. X 2 times), followed by water (3.0 vol.). Dichloromethane layer was concentrated under vacuum at 43±2°C up to 4.0 volume stage. Acetone (4.0 vol.) was added to the reaction mass. This reaction mass was added dropwise to Cyclohexane (20.0 vol.) at 10 to 14 °C with constant stirring, the reaction mass was slowly warmed to 25 °C and continued to stir at the same temperature for 1 hour. Filtered and washed with Cyclohexane (5.0 vol.) and suck dried; dried in VTD at 60±5°C for 30 to 40 h under vacuum to yield 4-[4-[4-[4-[[ (3R,5R)-5- (2,4- difluorophenyl)tetrahydro-5(lH- 1 ,2,4-triazol- 1 -ylmethyl)-3-furanyl]methoxy]phenyl] - 1 - piperazinyl]phenyl]-2-[(lS,2S)-l-ethyl-2-hydroxy propyl] -2,4-dihydro-3H-l,2,4-triazol- 3- one of the structural formula (IV) of amorphous form with 95 % yield. Characteristic Physico-Chemical Data of Amorphous Form of the Compound of Structural Formula IV Physical appearance: Off-white to white solid X-ray Powder Diffraction Pattern: See Figure 6 DSC: See Figure 7
References:
WO2017/51342,2017,A1 Location in patent:Page/Page column 19; 20
![D-threo-Pentitol, 2,5-anhydro-1,3,4-trideoxy-2-C-(2,4-difluorophenyl)-4-[[4-[4-[4-[[[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-2-formylhydrazinyl]carbonyl]amino]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1H-1,2,4-triazol-1-yl)-](/CAS/20211123/GIF/1350560-57-4.gif)
1350560-57-4
2 suppliers
inquiry
171228-49-2
645 suppliers
$6.00/5mg
74853-08-0
254 suppliers
$81.00/5g
171228-49-2
645 suppliers
$6.00/5mg
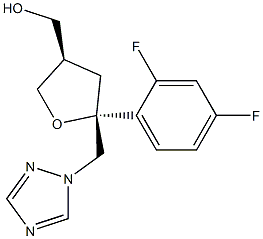
160709-02-4
62 suppliers
inquiry
171228-49-2
645 suppliers
$6.00/5mg

454479-36-8
2 suppliers
inquiry
171228-49-2
645 suppliers
$6.00/5mg