概述[1]
阿巴卡韦,化学名称为(1S,4R)-4-[2-氨基-6-(环丙氨基)-9H-嘌呤-9-基]-2-环戊烯基-1-甲醇。它属于核苷类逆转录酶抑制剂抗艾滋病毒药物,为HIV-1RT底物的竞争性抑制剂,抑制RT活性,阻碍前病毒DNA合成,并由于在结构上3’缺乏羟基,当它们结合到前病毒DNA链的3’末端时,不能再进行5’至3’磷酸二酯键的结合,终止了病毒DNA链的延长。通过上述作用机制,抑制HIV复制,并且与HIV-1RT亲和力远比与细胞内正常DNA聚合酶亲和力强,因此具有一定的治疗指数。
药理作用[2]
本品是一个新的碳环2'-脱氧鸟苷核苷类药物,其口服生物利用度高,易渗入中枢神经系统。与其他核苷类反转录酶抑制剂一样,它是一个无活性的前药,在体内经四个步骤代谢成为具活性的三磷酸酯,并通过以下两条途径发挥抑制人免疫缺陷病毒(HIV)反转录酶的作用:①竞争性地抑制2'-脱氧鸟苷三磷酸酯(dGTP)(DNA合成片段之一)结合进入核酸链。②通过阻止新碱基的加入而有效地终止DNA链的合成。
适应证[2]
与其他抗艾滋病药物联合应用,治疗HIV感染的成年患者及3个月以上儿童患者。
不良反应[2]
主要有恶心、呕吐、不适及疲劳,口服液有轻微的胃肠道反应。严重者也可伴有肝衰、肾衰、低血压,甚至死亡。
禁忌[2]
对本品制剂中任何成分过敏的病人禁用本品。禁用于严重肝功能受损的病人及终末期肾病患者。
制备[3]
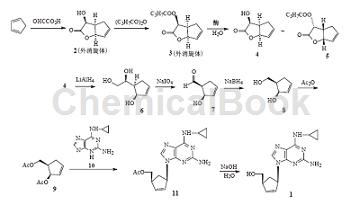
(±)-4-endo-4-羟基-2-氧杂二环[3.3.0]辛-7-烯-3-酮(2)
1L锥形瓶中加入50%乙醛酸溶液(296g,2mol)和水(440ml),冰浴冷却下加入环戊二烯(145.2g,2.2mol),0~5℃搅拌反应56h。反应液用正庚烷洗涤,加氯化钠充分饱和后,用二氯甲烷萃取,二氯甲烷相用饱和碳酸氢钠溶液洗涤,干燥蒸去溶剂,剩余的浅棕色油状物用乙醚重结晶,烘干,得白色固体2(89.6g,32%)(文献:收率31.2%),mp68~70℃。1HNMR与文献一致。
(±)-4-endo-4-丁酰氧基-2-氧杂二环[3.3.0]辛-7-烯-3-酮(3)
2(231.5g,1.65mol)、碳酸氢钾(314.2g,3.14mol)、丁酸酐(282.5g,1.79mol)和乙腈(2.3L)室温搅拌17h,滤除不溶物,滤液蒸除溶剂,剩余物中加入乙酸乙酯,用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,过滤,滤液蒸除溶剂,得浅棕色油状3(348g,粗收率100%)。直接投入下步反应。
(1R,4S,5R)-(–)-4-endo-4-羟基-2-氧杂二环-[3.3.0]辛-7-烯-3-酮(4)
3(97g,0.46mol)和0.1mol/L磷酸二氢钾溶液(800ml)置于反应瓶中,室温搅拌下加入10mol/L氢氧化钠溶液调至pH6.6,加入脂肪酶AmanoPS(2.9g),室温搅拌,用10mol/L氢氧化钠溶液控制反应体系pH6.55~6.65,约10h反应完毕。加入氯化
钠(100g),反应液离心分离,上清液用乙酸乙酯萃取,用无水硫酸钠干燥,过滤,滤液蒸除溶剂,剩余物置冰箱静置析晶,过滤,滤饼用二氯甲烷重结晶,得类白色固体4(18.1g,56%),mp94~96℃,[α]D20–102.4°(c1,氯仿)[文献:收率60%,[α]D20–94°(c1,氯仿)]。1HNMR与文献一致。
(1R,5S,1'S)-5-(1',2'-二羟基乙基)环戊-2-烯-1-醇(6)
氮气保护下用LiAlH4(16.4g,0.431mol)在无水THF(275ml)中回流0.5h后,滴加4(55g,0.392mol)的THF溶液(275ml),1.5h内加完。滴加15%氢氧化钠溶液(16.4ml),搅拌10mn,过滤,滤饼用THF洗涤,洗液和滤液合并后蒸干,得褐色油状6(54.2g),直接用于下步反应。
(1R,2R)-2-羟基环戊-3-烯-1-甲醇(8)
如上所得6溶于水(540ml)中,加入乙醚(270ml),0℃搅拌下分批加入高碘酸钠(85.1g,0.398mol),1.5h内加完,0℃搅拌2h后加入乙二醇(3.6ml),继续搅拌1h,过滤,滤液蒸除乙醚,剩余物中加入乙醇(140ml),冷却至0℃,分批加入硼氢化钠(15.1g,0.398mol),同温搅拌1h。减压蒸除乙醇,残留物用乙酸乙酯萃取,萃取相用饱和盐水洗涤,无水硫酸钠干燥,过滤,滤液蒸除溶剂,得黄色油状8(26.8g,3步连乘收率60%),[α]D20–128.0°(c1,氯仿)[文献:3步收率52%,[α]D20–121°(c1,氯仿)]。1HNMR与文献一致。
(1R,2R)-2-乙酰氧基环戊-3-烯-1-甲醇乙酸酯(9)
8(25g,0.219mol)溶于二氯甲烷(250ml)中,0℃下加入三乙胺(48.7g,0.482mol),氮气保护下滴加乙酐(67g,0.657mol),再加入DMAP(0.25g,2.0mmol),同温搅拌2h。加5%盐酸调至pH2,静置分层,二氯甲烷层依次用饱和碳酸氢钠溶液和饱和盐水洗涤,无水硫酸钠干燥,过滤,滤液蒸除溶剂,得黄色油状9(42.5g,98%),[α]D20–180.8°(c1,二氯甲烷)[文献:收率90%,[α]D20–178.0°(c1,二氯甲烷)]。1HNMR与文献一致。
2-氨基-6-环丙胺基-9H-嘌呤(10)
2-氨基-6-氯-9H-嘌呤(40g,0.236mol)、环丙胺(80.7g,1.416mol)和甲醇(200ml)加热至回流反应17h,冰浴冷却下加入碳酸钾(40g,0.290mol),搅拌2h,过滤,滤液浓缩至有固体析出,冰箱静置过夜。过滤,滤饼用甲醇重结晶,得10(39.4g,88%),mp149~152℃(文献:收率78%,mp150~152℃)。1HNMR与文献一致。
阿巴卡韦(1)
氮气保护下将60%NaH(3.2g,0.08mol)置于THF(190ml)中,45℃加入10(12.55g,0.066mol),同温搅拌1h,加入四(三苯基膦)钯(7.7g,6.6mmol)和9(14.5g,0.073mol),室温搅拌60h。减压蒸除THF,剩余物中加入二氯甲烷(300ml)和水(300ml),过滤,滤液静置分层,水层用二氯甲烷萃取,合并二氯甲烷层,用饱和盐水洗2次,浓缩至约100ml后用5%盐酸提取,合并水层,加碳酸钾调至pH9,用乙酸乙酯萃取,萃取相用饱和盐水洗2次,无水硫酸钠干燥,过滤,滤液蒸除溶剂,残余物中加入1mol/L氢氧化钠溶液(132ml)和乙醇(45ml),室温搅拌8h,用乙酸乙酯萃取,萃取相用饱和盐水洗涤,无水硫酸钠干燥,过滤,滤液蒸除溶剂,剩余物用甲醇-丙酮重结晶,得1(6.0g,2步收率32%)(文献:48%),mp155~160℃(产物为泡沫状粉末,难以准确测得熔点),[α]D20–52.9°(c1,甲醇)[文献(TheMerckIndex13th,No.1):mp165℃,
主要参考资料
[1] CN201410027146.5一种阿巴卡韦的制备方法
[2] 新编临床药物学
[3] 阿巴卡韦的合成